Citation: George L. Mendz, Nadeem O. Kaakoush, Julie A. Quinlivan. New techniques to characterise the vaginal microbiome in pregnancy[J]. AIMS Microbiology, 2016, 2(1): 55-68. doi: 10.3934/microbiol.2016.1.55

| [1] | Lawn JE, Cousens S, Zupan J (2005) 4 million neonatal deaths: When? Where? Why? Lancet 365: 891–900. |
| [2] |
Goldenberg RL, Culhane JF, Iams JD, et al. (2008) Preterm birth 1: Epidemiology and causes of preterm birth. Lancet 371: 75–84. doi: 10.1016/S0140-6736(08)60074-4

|
| [3] |
Shatrov JG, Birch SC, Lam, LT, et al. (2010) Chorioamnionitis and cerebral palsy. Obstet Gynecol 116: 387–392. doi: 10.1097/AOG.0b013e3181e90046
|
| [4] | Hussein J, Ugwumadu A, Witkin SS (2011) Editor’s choice. Brit J Obst Gynaecol 118: i–ii. |
| [5] |
Conti N, Torricelli M, Voltolini C, et al. (2015) Term histologic chorioamnionitis: a heterogeneous condition. Eur J Obstet Gynecol Reprod Biol 188: 34–38. doi: 10.1016/j.ejogrb.2015.02.034
|
| [6] | Pretorius C, Jagatt A, Lamont RF (2007) The relationship between periodontal disease, bacterial vaginosis, and preterm birth. J Perinat Med 35: 93–99. |
| [7] |
Romero R, Espinoza J, Chaiworapongsa T, et al. (2002) Infection and prematurity and the role of preventive strategies. Semin Neonatol 7: 259–274. doi: 10.1016/S1084-2756(02)90121-1
|
| [8] |
Romero R, Mazor M (1988) Infection and preterm labor. Clin Obstet Gynecol 31: 553–584. doi: 10.1097/00003081-198809000-00006
|
| [9] | Smaill F (2001) Antibiotics for asymptomatic bacteriuria in pregnancy. Chocrane Database Syst. Rev 2: CD000490. |
| [10] |
Lopez NJ, Uribe S, Martinez, B (2015) Effect of periodontal treatment on pretern birth: a systematic review of meta-analyses. Periodontol 2000 67: 87–130. doi: 10.1111/prd.12073
|
| [11] |
Aagard K, Riehle K, Ma J, et al. (2012) A Metagenomic approach to characterization of the vaginal microbiome signature in pregnancy. PLoS One 7: e36466. doi: 10.1371/journal.pone.0036466
|
| [12] | Relman D (2012) Learning about who we are. Nature 468: 194–195. |
| [13] |
Eschenbach DA, Thwin SS, Patton DL, et al. (2000) Influence of the normal menstrual cycle on vaginal tissue, discharge, and microflora. Clin Infect Dis 30: 901–907. doi: 10.1086/313818
|
| [14] |
Burton JP, Reid G (2002) Evaluation of the bacterial vaginal flora of 20 postmenopausal women by direct (Nugent score) and molecular (polymerase chain reaction and denaturing gradient gel electrophoresis) techniques. J Infect Dis 186: 1770–1780. doi: 10.1086/345761
|
| [15] |
Clarke JG, Peipert JF, Hillier SL, et al. (2002) Microflora changes with the use of vaginal microbicide. Sex Transm Dis 29: 288–293. doi: 10.1097/00007435-200205000-00007
|
| [16] |
Ness RB, Hillier SL, Kip KE, et al. (2005) Douching, pelvic inflammatory disease, and incident gonococcal and chlamydial genital infection in a cohort of high-risk women. Am J Epidemiol 161: 186–195. doi: 10.1093/aje/kwi025
|
| [17] | Wilson M (2005) Microbial inhabitants of humans: their ecology and role in health and disease. Cambridge: University Press, UK; 206–250. |
| [18] |
Choi SJ, Park SD, Jang IH, et al. (2012) The prevalence of vaginal microorganisms in pregnant women with preterm labor and preterm birth. Ann Lab Med 32: 194–200. doi: 10.3343/alm.2012.32.3.194
|
| [19] | Giraldo PC, Araujo ED, Junior, JE, et al. (2012) The prevalence of urogenital infections in pregnant women experiencing preterm and full-term labor. Inf Dis Obst Gynecol 2012: 878241. |
| [20] |
Wen A, Srinivasan U, Goldber D, et al. (2014) Selected vaginal bacteria and risk of preterm birth: An ecological perspective. J Infect Dis 209: 1087–1094. doi: 10.1093/infdis/jit632
|
| [21] | Ravel J, Gajer P, Abdo Z, et al. (2011). Vaginal microbiome of reproductive-age women. Proc Natl Acad Sci U S A 108 (Suppl 1): 4680–4687. |
| [22] |
Srinivasan S, Hoffman NG, Morgan MT, et al. (2012) Bacterial communities in women with bacterial vaginosis: high resolution phylogenetic analyses reveal relationships of microbiota to clinical criteria. PLoS One 7: e37818. doi: 10.1371/journal.pone.0037818
|
| [23] | Fettweis JM, Serrano MG, Sheth NU, et al. (2012) Species-level classification of the vaginal microbiome. BMC Genomics 13 (Suppl 8): S17. |
| [24] | Ma CX, Ellis M J (2013) The cancer genome atlas: clinical applications for breast cancer. Oncology 27: 1263-1269, 1274–1279. |
| [25] | Pinto R, De Summa S, Petriella D, et al. (2014) The value of new high-throughput technologies for diagnosis and prognosis in solid tumors. Cancer Biomark 14: 103–117. |
| [26] |
Renkema KY, Stokman MF, Giles RH et al. (2014) Next-generation sequencing for research and diagnostics in kidney disease. Nat Rev Nephrol 10: 433–444. doi: 10.1038/nrneph.2014.95
|
| [27] |
Stadler ZK, Schrader KA, Vijai J, et al. (2014) Cancer genomics and inherited risk. J Clin Oncol 32: 687–698. doi: 10.1200/JCO.2013.49.7271
|
| [28] |
Huang YE, Wang Y, He, Ji, et al. (2015) Homogeneity of the vaginal microbiome at the cervix, posterior fornix, and vaginal canal in pregnant Chinese women. Microb Ecol 69: 407–414. doi: 10.1007/s00248-014-0487-1
|
| [29] |
Hyman RW, Fukushima M, Jiang H, et al. (2014) Diversity of the vaginal microbiome correlates with preterm birth. Reprod Sci 21: 32–40. doi: 10.1177/1933719113488838
|
| [30] |
Cox MJ, Cookson WO, Moffatt MF (2013) Sequencing the human microbiome in health and disease. Hum Mol Genet 22: R88–R94. doi: 10.1093/hmg/ddt398
|
| [31] | Quince C, Lanzen A, Davenport RJ, et al. (2011) Removing noise from pyrosequenced amplicons. BMC Bioinformatics 6: 639–641. |
| [32] | Shah N, Tang H, Doak TG, et al. (2011) Comparing bacterial communities inferred from 16S rRNA gene sequencing and shotgun metagenomics. Pac Symp Biocomput 165–176. |
| [33] |
Brooks JP, Edwards DJ, Harwich, et al. (2015) The truth about metagenomics: quantifying and counteracting bias in 16S rRNA studies. BMC Microbiol 15: 66. doi: 10.1186/s12866-015-0351-6
|
| [34] | Starke IC, Vahjen W, Pieper R, et al. (2014) The Influence of DNA extraction procedure and primer set on the bacterial community analysis by pyrosequencing of barcoded 16S rRNA gene amplicons. Mol Biol Int 2014: 548683. |
| [35] |
Klindworth A, Pruesse E, Schweer T, et al. (2013) Evaluation of general 16S ribosomal RNA gene PCR primers for classical and next-generation sequencing-based diversity studies. Nucleic Acids Res 41: e1. doi: 10.1093/nar/gks808
|
| [36] | Nelaakanta G, Sultana H (2013) The use of metagenomics approaches to analyse changes in microbial communities. Microbiol Insights 6: 37–48. |
| [37] |
Schloss PD, Westcott SL, Ryabin T, et al. (2009) Introducing mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl Environ Microbiol 75: 7537–7541. doi: 10.1128/AEM.01541-09
|
| [38] |
Caporaso JG, Kuczynski J, Stombaugh J, et al. (2010) QIIME allows analysis of high-throughput community sequencing data. Nat Methods 7: 335–336. doi: 10.1038/nmeth.f.303
|
| [39] |
Edgar RC (2013) UPARSE: highly accurate OTU sequences from microbial amplicon reads. Nat Methods 10: 996–998. doi: 10.1038/nmeth.2604
|
| [40] | Links MG, Chaban B, Hemmingsen SM, et al. (2013) mPUMA: a computational approach to microbiota analysis by de novo assembly of operational taxonomic units based on protein-coding barcode sequences. Microbiome 15: 23. doi: 10.1186/2049-2618-2-23. |
| [41] |
Fettweis JM, Serrano MG, Girerd PH, et al. (2012) A new era of the vaginal microbiome: Advances using next-generation sequencing. Chem Biodiver 9: 965–976. doi: 10.1002/cbdv.201100359
|
| [42] |
Meyer F, Paarmann D, D'Souza M, et al. (2008) The metagenomics RAST server - A public resource for the automatic phylogenetic and functional analysis of metagenomes. BMC Bioinformatics 9: 386. doi: 10.1186/1471-2105-9-386
|
| [43] |
Zakrzewski M, Bekel T, Ander C, et al. (2013) MetaSAMS--a novel software platform for taxonomic classification, functional annotation and comparative analysis of metagenome datasets. J Biotechnol 167: 156–165. doi: 10.1016/j.jbiotec.2012.09.013
|
| [44] |
Hunter S, Corbett M, Denise H, et al. (2014) EBI metagenomics - a new resource for the analysis and archiving of metagenomic data. Nucleic Acids Res 42: D600–D606. doi: 10.1093/nar/gkt961
|
| [45] | Anderson MJ (2001) A new method for non‐parametric multivariate analysis of variance. Austral J Ecol 26: 32–46. |
| [46] |
Clarke KR (1993) Non-parametric multivariate analyses of changes in community structure. Austr J Ecol 18: 117–143. doi: 10.1111/j.1442-9993.1993.tb00438.x
|
| [47] | Ganu RS, Ma J, & Aagaard KM (2013) The role of microbial communities in parturition: is there evidence of association with preterm birth and perinatal morbidity and mortality? Am J Perinatol 30: 613–624. |
| [48] |
Walther-Antonio MRS, Jeraldo, Miller MEB, Yeoman, et al. (2014) Pregnancy’s stronghold on the vaginal microbiome. PLOS One 9: e98514. doi: 10.1371/journal.pone.0098514
|
| [49] |
Romero R, Hassan SS, Gajer P, et al. (2014) The vaginal microbiota of pregnant women who subsequently have spontaneous preterm labor and delivery and those with a normal delivery at term. Microbiome 2: 18. doi: 10.1186/2049-2618-2-18
|
| [50] |
Jespers V, Menten J, Smet H, et al. (2012) Quantification of bacterial species of the vaginal microbiome in different groups of women, using nucleic acid amplification tests. BMC Microbiology 12: 83. doi: 10.1186/1471-2180-12-83
|
| [51] |
Romero R, Hassan SS, Gajer P, et al. (2014) The composition and stability of the vaginal microbiota of normal pregnant women is different from that of non-pregnant women. Microbiome 2: 4. doi: 10.1186/2049-2618-2-4
|
| [52] |
Witkin SS (2015) The vaginal microbiome, vaginal anti-microbial defence mechanisms and the clinical challenge of reducing infection-related preterm birth. Brit J Obst Gynaecol 122: 213–218. doi: 10.1111/1471-0528.13115
|
| [53] |
Mysorekar IU, Cao B (2014) Microbiome in parturition and preterm birth. Semin Reprod Med 32: 50–55. doi: 10.1055/s-0033-1361830
|
| [54] |
Ling Z, Kong J, Liu F, et al. (2010) Molecular analysis of the diversity of vaginal microbiota associated with bacterial vaginosis. BMC Genomics 11: 488. doi: 10.1186/1471-2164-11-488
|
| [55] |
Africa CWJ, Nel J, Stemmet, M (2014) Anaerobes and bacterial vaginosis in pregnancy: Virulence factors contributing to vaginal colonization. Int J Environ Res Public Health 11: 6979–7000. doi: 10.3390/ijerph110706979
|
| [56] |
Martin DH (2012) The microbiota of the vagina and its influence on women’s health and disease. Am J Med Sci 343: 2–9. doi: 10.1097/MAJ.0b013e31823ea228
|
| [57] | Mendz GL, Petersen R, Quinlivan JA, et al. (2014) Potential involvement of Campylobacter curvus and Haemophilus parainfluenzae in preterm birth. Br Med J Case Rep 2014. doi: 10.1136/bcr-2014-205282. |
| [58] | Kaakoush NO, Quilivan JA, Mendz, GL (2014) Bacteroides and Hafnia Infections associated with chorioamnionitis and preterm birth. J Clin Gynecol Obstet 3: 76–79. |
| [59] |
Quinlivan JA, Kaakoush NO, & Mendz GL (2014) Acinetobacter species associated with spontaneous preterm birth and histological chorioamnionitis. Br J Med Med Res 4: 5293–5297. doi: 10.9734/BJMMR/2014/12004
|
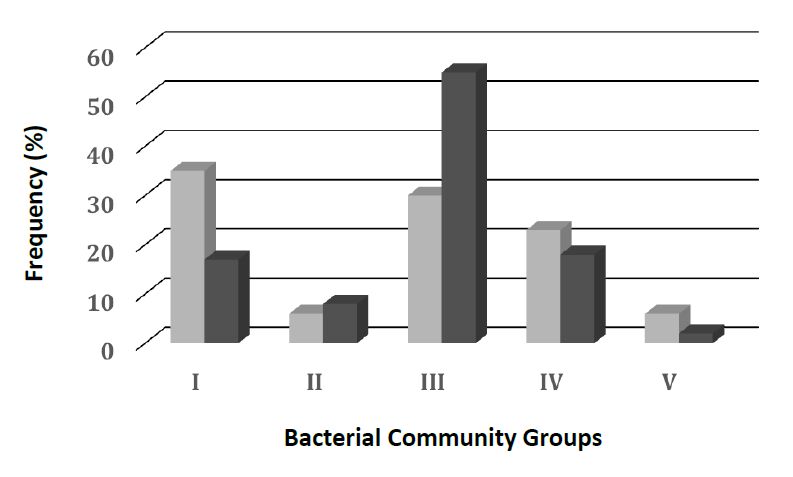
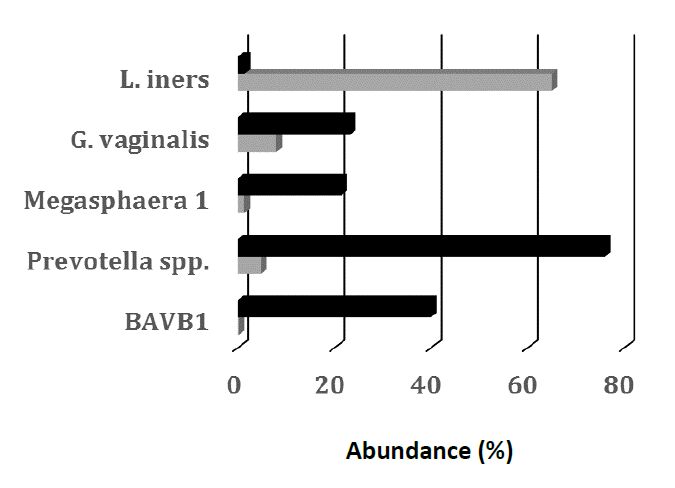
Figures(2) / Tables(1)

George L. Mendz, Nadeem O. Kaakoush, Julie A. Quinlivan. New techniques to characterise the vaginal microbiome in pregnancy[J]. AIMS Microbiology, 2016, 2(1): 55-68. doi: 10.3934/microbiol.2016.1.55







 DownLoad:
DownLoad: