Citation: Dawn B. Goldsmith, Zoe A. Pratte, Christina A. Kellogg, Sara E. Snader, Koty H. Sharp. Stability of temperate coral Astrangia poculata microbiome is reflected across different sequencing methodologies[J]. AIMS Microbiology, 2019, 5(1): 62-76. doi: 10.3934/microbiol.2019.1.62

| [1] | Peters EC, Cairns SD, Pilson ME, et al. (1988) Nomenclature and biology of Astrangia poculata (= A. danae,= A. astreiformis)(Cnidaria: Anthozoa). P Biol Soc Wash 101: 234–250. |
| [2] |
Dimond J, Kerwin A, Rotjan R, et al. (2013) A simple temperature-based model predicts the upper latitudinal limit of the temperate coral Astrangia poculata. Coral Reefs 32: 401–409. doi: 10.1007/s00338-012-0983-z

|
| [3] | LaJeunesse TC, Parkinson JE, Reimer JD (2012) A genetics-based description of Symbiodinium minutum sp. nov. and S. psygmophilum sp. nov.(Dinophyceae), two dinoflagellates symbiotic with cnidaria. J Phycol 48: 1380–1391. |
| [4] |
Thornhill DJ, Kemp DW, Bruns BU, et al. (2008) Correspondence between cold tolerance and temperate biogeography in a Western Atlantic symbiodinium (Dinophyta) lineage 1. J Phycol 44: 1126–1135. doi: 10.1111/j.1529-8817.2008.00567.x
|
| [5] |
Sharp KH, Pratte ZA, Kerwin AH, et al. (2017) Season, but not symbiont state, drives microbiome structure in the temperate coral Astrangia poculata. Microbiome 5: 120. doi: 10.1186/s40168-017-0329-8
|
| [6] |
LaJeunesse TC, Parkinson JE, Gabrielson PW, et al. (2018) Systematic revision of Symbiodiniaceae highlights the antiquity and diversity of coral endosymbionts. Curr Biol 28: 2570–2580. doi: 10.1016/j.cub.2018.07.008
|
| [7] |
Dimond J, Carrington E (2007) Temporal variation in the symbiosis and growth of the temperate scleractinian coral Astrangia poculata. Mar Ecol Prog Ser 348: 161–172. doi: 10.3354/meps07050
|
| [8] |
Shade A, Handelsman J (2012) Beyond the Venn diagram: the hunt for a core microbiome. Environ Microbiol 14: 4–12. doi: 10.1111/j.1462-2920.2011.02585.x
|
| [9] |
Hernandez-Agreda A, Gates RD, Ainsworth TD (2017) Defining the core microbiome in corals' microbial soup. Trends Microbiol 25: 125–140. doi: 10.1016/j.tim.2016.11.003
|
| [10] |
Ainsworth TD, Krause L, Bridge T, et al. (2015) The coral core microbiome identifies rare bacterial taxa as ubiquitous endosymbionts. ISME J 9: 2261–2274. doi: 10.1038/ismej.2015.39
|
| [11] | Edwards U, Rogall T, Blöcker H, et al. (1989) Isolation and direct complete nucleotide determination of entire genes. Characterization of a gene coding for 16S ribosomal RNA. Nucleic Acids Res 17: 7843–7853. |
| [12] | Stackebrandt E, Liesack W (1993) Nucleic acids and classification, In: Goodfellow M, O'Donnell AG, Editors, Handbook of New Bacterial Systematics, England: Academic Press, 152–189. |
| [13] |
DeLong EF (1992) Archaea in coastal marine environments. P Natl Acad Sci USA 89: 5685–5689. doi: 10.1073/pnas.89.12.5685
|
| [14] |
Caporaso JG, Kuczynski J, Stombaugh J, et al. (2010) QIIME allows analysis of high-throughput community sequencing data. Nat Methods 7: 335–336. doi: 10.1038/nmeth.f.303
|
| [15] |
Werner JJ, Koren O, Hugenholtz P, et al. (2012) Impact of training sets on classification of high-throughput bacterial 16s rRNA gene surveys. ISME J 6: 94. doi: 10.1038/ismej.2011.82
|
| [16] |
McDonald D, Price MN, Goodrich J, et al. (2012) An improved Greengenes taxonomy with explicit ranks for ecological and evolutionary analyses of bacteria and archaea. ISME J 6: 610–618. doi: 10.1038/ismej.2011.139
|
| [17] |
DeSantis TZ, Hugenholtz P, Larsen N, et al. (2006) Greengenes, a chimera-checked 16S rRNA gene database and workbench compatible with ARB. Appl Environ Microb 72: 5069–5072. doi: 10.1128/AEM.03006-05
|
| [18] |
Edgar RC (2010) Search and clustering orders of magnitude faster than BLAST. Bioinformatics 26: 2460–2461. doi: 10.1093/bioinformatics/btq461
|
| [19] | Edgar R (2016) UCHIME2: improved chimera prediction for amplicon sequencing. BioRxiv: 074252. |
| [20] | Goldsmith DB, Kellogg CA, Cold-water coral microbiomes (Astrangia poculata) from Narrangansett Bay: sequence data. U.S. Geological Survey Data Release, 2019. Available from: https://doi.org/10.5066/P9C2XCQQ. |
| [21] | Bolyen E, Rideout JR, Dillon MR, et al. (2018) QIIME 2: Reproducible, interactive, scalable, and extensible microbiome data science. PeerJ Preprints: 2167–9843. |
| [22] | Oksanen J, Blanchet FG, Friendly M, et al. (2019) vegan: Community Ecology Package. R package version 2.5-4: https://CRAN.R-project.org/package=vegan. |
| [23] | R Core Team (2015) R: A language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria. |
| [24] |
Clarke KR (1993) Non-parametric multivariate analyses of changes in community structure. Aust J Ecol 18: 117–143. doi: 10.1111/j.1442-9993.1993.tb00438.x
|
| [25] |
Bray JR, Curtis JT (1957) An ordination of the upland forest communities of southern Wisconsin. Ecol Monogr 27: 325–349. doi: 10.2307/1942268
|
| [26] | Anderson MJ (2001) A new method for non-parametric multivariate analysis of variance. Austral Ecol 26: 32–46. |
| [27] | Nawrocki EP (2009) Structural RNA homology search and alignment using covariance models, Ph.D. thesis, Washington University in St. Louis School of Medicine. |
| [28] |
Price MN, Dehal PS, Arkin AP (2010) FastTree 2-Approximately maximum-likelihood trees for large alignments. PLoS One 5: e9490. doi: 10.1371/journal.pone.0009490
|
| [29] |
Paradis E, Claude J, Strimmer K (2004) APE: analyses of phylogenetics and evolution in R language. Bioinformatics 20: 289–290. doi: 10.1093/bioinformatics/btg412
|
| [30] |
Altschul SF, Gish W, Miller W, et al. (1990) Basic local alignment search tool. J Mol Biol 215: 403–410. doi: 10.1016/S0022-2836(05)80360-2
|
| [31] |
Shannon C (1948) A mathematical theory of communication. Bell Syst Tech J 27: 379–423. doi: 10.1002/j.1538-7305.1948.tb01338.x
|
| [32] |
Hutcheson K (1970) A test for comparing diversities based on the Shannon formula. J Theor Biol 29: 151–154. doi: 10.1016/0022-5193(70)90124-4
|
| [33] |
D'Amore R, Ijaz UZ, Schirmer M, et al. (2016) A comprehensive benchmarking study of protocols and sequencing platforms for 16S rRNA community profiling. BMC Genomics 17: 55. doi: 10.1186/s12864-015-2194-9
|
| [34] |
Fouhy F, Clooney AG, Stanton C, et al. (2016) 16S rRNA gene sequencing of mock microbial populations-impact of DNA extraction method, primer choice and sequencing platform. BMC Microbiol 16: 123. doi: 10.1186/s12866-016-0738-z
|
| [35] |
Smith BC, McAndrew T, Chen Z, et al. (2012) The cervical microbiome over 7 years and a comparison of methodologies for its characterization. PLoS One 7: e40425. doi: 10.1371/journal.pone.0040425
|
| [36] |
Caporaso JG, Lauber CL, Walters WA, et al. (2011) Global patterns of 16S rRNA diversity at a depth of millions of sequences per sample. P Natl Acad Sci USA 108: 4516–4522. doi: 10.1073/pnas.1000080107
|
| [37] | Tremblay J, Singh K, Fern A, et al. (2015) Primer and platform effects on 16S rRNA tag sequencing. Front Microbiol 6: 771. |
| [38] |
Nelson MC, Morrison HG, Benjamino J, et al. (2014) Analysis, optimization and verification of Illumina-generated 16S rRNA gene amplicon surveys. PLoS One 9: e94249. doi: 10.1371/journal.pone.0094249
|
| [39] |
Gihring TM, Green SJ, Schadt CW (2012) Massively parallel rRNA gene sequencing exacerbates the potential for biased community diversity comparisons due to variable library sizes. Environ Microbiol 14: 285–290. doi: 10.1111/j.1462-2920.2011.02550.x
|
| [40] |
Schirmer M, D'Amore R, Ijaz UZ, et al. (2016) Illumina error profiles: resolving fine-scale variation in metagenomic sequencing data. BMC Bioinformatics 17: 125. doi: 10.1186/s12859-016-0976-y
|
| [41] |
Huse SM, Welch DM, Morrison HG, et al. (2010) Ironing out the wrinkles in the rare biosphere through improved OTU clustering. Environ Microbiol 12: 1889–1898. doi: 10.1111/j.1462-2920.2010.02193.x
|
| [42] |
Edgar RC (2017) Accuracy of microbial community diversity estimated by closed-and open-reference OTUs. PeerJ 5: e3889. doi: 10.7717/peerj.3889
|
| [43] | Kopylova E, Navas-Molina JA, Mercier C, et al. (2016) Open-source sequence clustering methods improve the state of the art. MSystems 1: e00003–00015. |
 microbiol-05-01-062-s001.xlsx microbiol-05-01-062-s001.xlsx |
 |
| microbiol-05-01-062-s002.xlsx |
|
| microbiol-05-01-062-s003.xlsx |
|
| microbiol-05-01-062-s004.xlsx |
|
| microbiol-05-01-062-s005.xlsx |
|
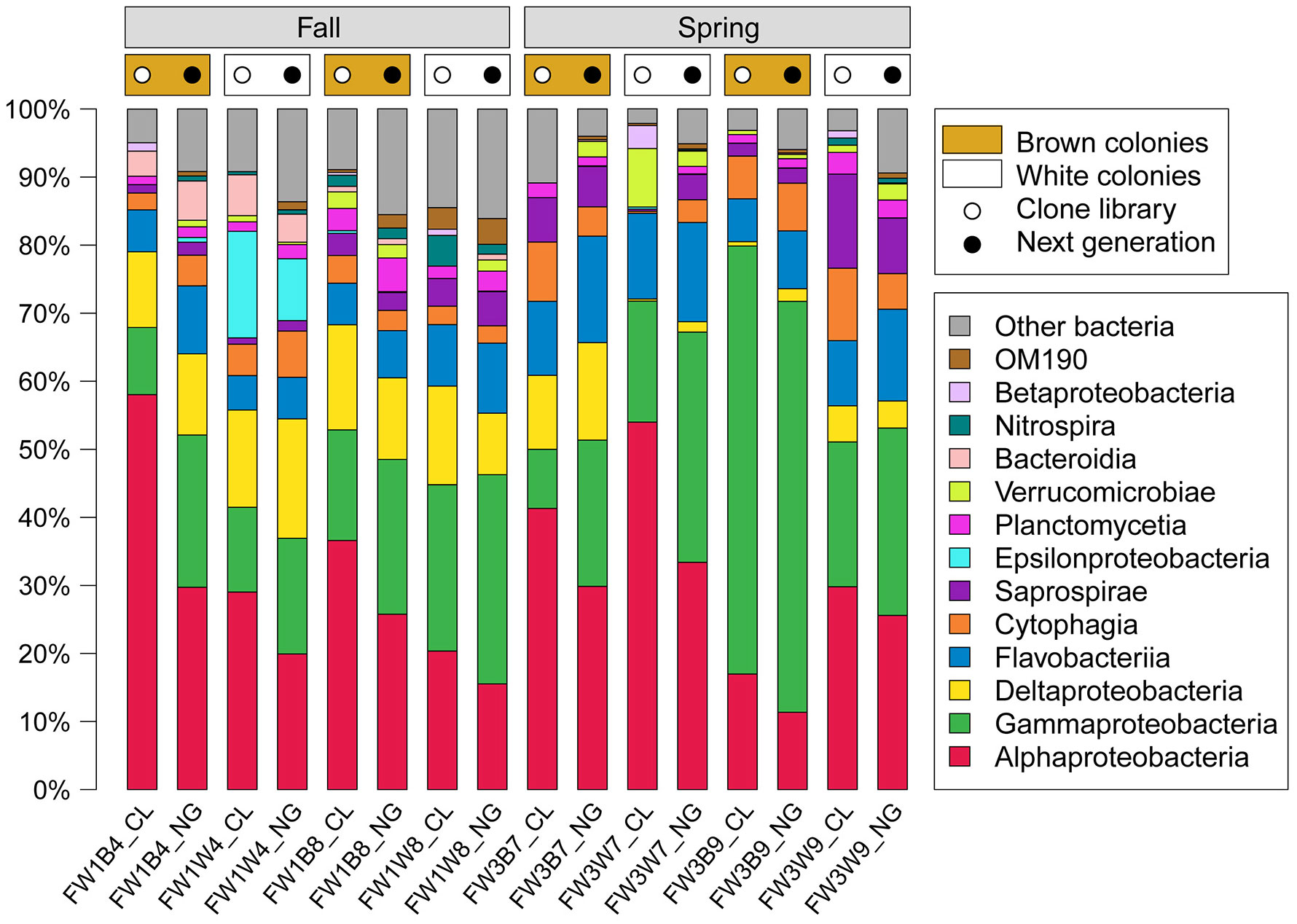
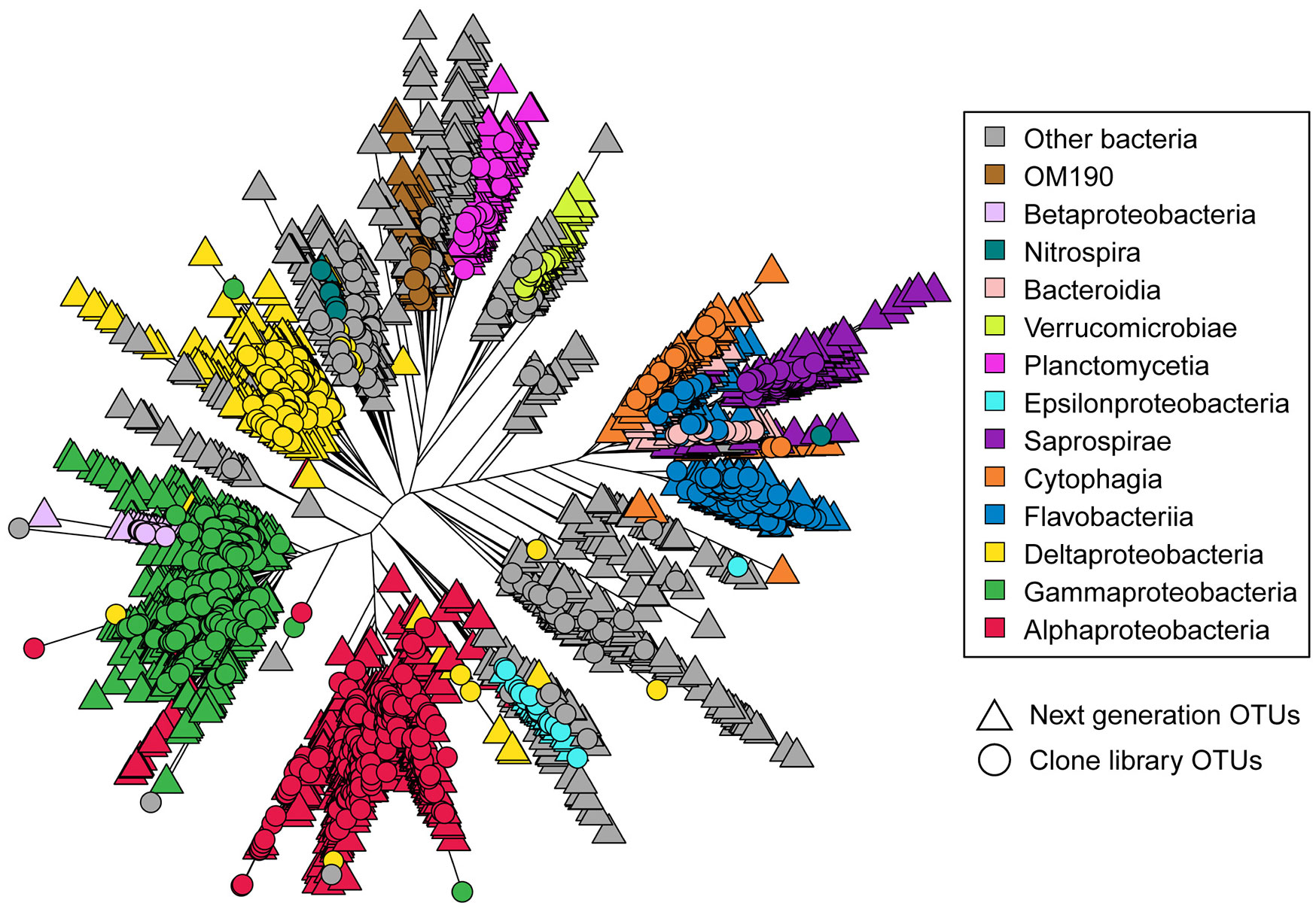
Figures(2) / Tables(4)

Dawn B. Goldsmith, Zoe A. Pratte, Christina A. Kellogg, Sara E. Snader, Koty H. Sharp. Stability of temperate coral Astrangia poculata microbiome is reflected across different sequencing methodologies[J]. AIMS Microbiology, 2019, 5(1): 62-76. doi: 10.3934/microbiol.2019.1.62







 DownLoad:
DownLoad: