Citation: Byron Carpenter. Current applications of mini G proteins to study the structure and function of G protein-coupled receptors[J]. AIMS Bioengineering, 2018, 5(4): 209-225. doi: 10.3934/bioeng.2018.4.209

| [1] |
Wise A, Gearing K, Rees S (2002) Target validation of G-protein coupled receptors. Drug Discov Today 7: 235–246. doi: 10.1016/S1359-6446(01)02131-6

|
| [2] |
Cherezov V, Rosenbaum DM, Hanson MA, et al. (2007) High-resolution crystal structure of an engineered human beta2-adrenergic G protein-coupled receptor. Science 318: 1258–1265. doi: 10.1126/science.1150577
|
| [3] |
Serrano-Vega MJ, Magnani F, Shibata Y, et al. (2008) Conformational thermostabilization of the beta1-adrenergic receptor in a detergent-resistant form. Proc Natl Acad Sci USA 105: 877–882. doi: 10.1073/pnas.0711253105
|
| [4] | Caffrey M (2015) A comprehensive review of the lipid cubic phase or in meso method for crystallizing membrane and soluble proteins and complexes. Acta Crystallogr F Struct Biol Commun 71: 3–18. |
| [5] |
Landau EM, Rosenbusch JP (1996) Lipidic cubic phases: A novel concept for the crystallization of membrane proteins. Proc Natl Acad Sci USA 93: 14532–14535. doi: 10.1073/pnas.93.25.14532
|
| [6] | Chan W, Said M, Zhang C, et al. (2018) GPCR-EXP: A semi-manually curated database for experimentally-solved and predicted GPCR structures. Available from: https://zhanglab.ccmb.med.umich.edu/GPCR-EXP/. |
| [7] |
Jazayeri A, Dias JM, Marshall FH (2015) From G Protein-coupled receptor structure resolution to rational drug design. J Biol Chem 290: 19489–19495. doi: 10.1074/jbc.R115.668251
|
| [8] |
Carpenter B, Tate CG (2017) Active state structures of G protein-coupled receptors highlight the similarities and differences in the G protein and arrestin coupling interfaces. Curr Opin Struct Biol 45: 124–132. doi: 10.1016/j.sbi.2017.04.010
|
| [9] |
Rasmussen SG, Devree BT, Zou Y, et al. (2011) Crystal structure of the beta2 adrenergic receptor-Gs protein complex. Nature 477: 549–555. doi: 10.1038/nature10361
|
| [10] |
Kang Y, Zhou XE, Gao X, et al. (2015) Crystal structure of rhodopsin bound to arrestin by femtosecond X-ray laser. Nature 523: 561–567. doi: 10.1038/nature14656
|
| [11] |
Westfield GH, Rasmussen SG, Su M, et al. (2011) Structural flexibility of the G alpha s alpha-helical domain in the beta2-adrenoceptor Gs complex. Proc Natl Acad Sci USA 108: 16086–16091. doi: 10.1073/pnas.1113645108
|
| [12] |
Van EN, Preininger AM, Alexander N, et al. (2011) Interaction of a G protein with an activated receptor opens the interdomain interface in the alpha subunit. Proc Natl Acad Sci USA 108: 9420–9424. doi: 10.1073/pnas.1105810108
|
| [13] |
Scheerer P, Park JH, Hildebrand PW, et al. (2008) Crystal structure of opsin in its G-protein-interacting conformation. Nature 455: 497–502. doi: 10.1038/nature07330
|
| [14] |
Hamm HE, Deretic D, Arendt A, et al. (1988) Site of G protein binding to rhodopsin mapped with synthetic peptides from the alpha subunit. Science 241: 832–835. doi: 10.1126/science.3136547
|
| [15] |
Pardon E, Laeremans T, Triest S, et al. (2014) A general protocol for the generation of Nanobodies for structural biology. Nat Protoc 9: 674–693. doi: 10.1038/nprot.2014.039
|
| [16] | Carpenter B, Tate CG (2016) Engineering a minimal G protein to facilitate crystallisation of G protein-coupled receptors in their active conformation. Protein Eng Des Sel 29: 583–594. |
| [17] |
Blankenship E, Vahedi-Faridi A, Lodowski DT (2015) The high-resolution structure of activated opsin reveals a conserved solvent network in the transmembrane region essential for activation. Structure 23: 2358–2364. doi: 10.1016/j.str.2015.09.015
|
| [18] |
Choe HW, Kim YJ, Park JH, et al. (2011) Crystal structure of metarhodopsin II. Nature 471: 651–655. doi: 10.1038/nature09789
|
| [19] |
Deupi X, Edwards P, Singhal A, et al. (2012) Stabilized G protein binding site in the structure of constitutively active metarhodopsin-II. Proc Natl Acad Sci USA 109: 119–124. doi: 10.1073/pnas.1114089108
|
| [20] |
Park JH, Morizumi T, Li Y, et al. (2013) Opsin, a structural model for olfactory receptors? Angew Chem Int Ed Engl 52: 11021–11024. doi: 10.1002/anie.201302374
|
| [21] |
Singhal A, Ostermaier MK, Vishnivetskiy SA, et al. (2013) Insights into congenital stationary night blindness based on the structure of G90D rhodopsin. EMBO Rep 14: 520–526. doi: 10.1038/embor.2013.44
|
| [22] |
Standfuss J, Edwards PC, D'Antona A, et al. (2011) The structural basis of agonist-induced activation in constitutively active rhodopsin. Nature 471: 656–660. doi: 10.1038/nature09795
|
| [23] |
Huang W, Manglik A, Venkatakrishnan AJ, et al. (2015) Structural insights into micro-opioid receptor activation. Nature 524: 315–321. doi: 10.1038/nature14886
|
| [24] |
Kruse AC, Ring AM, Manglik A, et al. (2013) Activation and allosteric modulation of a muscarinic acetylcholine receptor. Nature 504: 101–106. doi: 10.1038/nature12735
|
| [25] |
Rasmussen SG, Choi HJ, Fung JJ, et al. (2011) Structure of a nanobody-stabilized active state of the beta(2) adrenoceptor. Nature 469: 175–180. doi: 10.1038/nature09648
|
| [26] |
Ring AM, Manglik A, Kruse AC, et al. (2013) Adrenaline-activated structure of beta2-adrenoceptor stabilized by an engineered nanobody. Nature 502: 575–579. doi: 10.1038/nature12572
|
| [27] |
Weichert D, Kruse AC, Manglik A, et al. (2014) Covalent agonists for studying G protein-coupled receptor activation. Proc Natl Acad Sci USA 111: 10744–10748. doi: 10.1073/pnas.1410415111
|
| [28] |
Manglik A, Kobilka BK, Steyaert J (2017) Nanobodies to study G protein-coupled receptor structure and function. Annu Rev Pharmacol Toxicol 57: 19–37. doi: 10.1146/annurev-pharmtox-010716-104710
|
| [29] |
Nehme R, Carpenter B, Singhal A, et al. (2017) Mini-G proteins: Novel tools for studying GPCRs in their active conformation. PLoS One 12: e0175642. doi: 10.1371/journal.pone.0175642
|
| [30] |
Carpenter B, Nehme R, Warne T, et al. (2016) Structure of the adenosine A(2A) receptor bound to an engineered G protein. Nature 536: 104–107. doi: 10.1038/nature18966
|
| [31] |
Hanzal-Bayer M, Renault L, Roversi P, et al. (2002) The complex of Arl2-GTP and PDE delta: From structure to function. EMBO J 21: 2095–2106. doi: 10.1093/emboj/21.9.2095
|
| [32] |
Sunahara RK, Tesmer JJ, Gilman AG, et al. (1997) Crystal structure of the adenylyl cyclase activator Gsalpha. Science 278: 1943–1947. doi: 10.1126/science.278.5345.1943
|
| [33] |
Spiegel AM, Jr BP, Butrynski JE, et al. (1991) The G protein connection: Molecular basis of membrane association. Trends Biochem Sci 16: 338–341. doi: 10.1016/0968-0004(91)90139-M
|
| [34] | Carpenter B, Tate CG (2017) Expression, purification and crystallisation of the adenosine A2A receptor bound to an engineered Mini G protein. Bio Protoc, 7. |
| [35] |
Wan Q, Okashah N, Inoue A, et al. (2018) Mini G protein probes for active G protein-coupled receptors (GPCRs) in live cells. J Biol Chem 293: 7466–7473. doi: 10.1074/jbc.RA118.001975
|
| [36] | Carpenter B, Tate CG (2017) Expression and purification of mini G proteins from escherichia coli. Bio Protoc 7: e2235. |
| [37] |
Garcia-Nafria J, Lee Y, Bai X, et al. (2018) Cryo-EM structure of the adenosine A2A receptor coupled to an engineered heterotrimeric G protein. Elife 7: e35946. doi: 10.7554/eLife.35946
|
| [38] |
Garcia-Nafria J, Nehme R, Edwards PC, et al. (2018) Cryo-EM structure of the serotonin 5-HT1B receptor coupled to heterotrimeric Go. Nature 558: 620–623. doi: 10.1038/s41586-018-0241-9
|
| [39] |
Tsai CJ, Pamula F, Nehmé R, et al. (2018) Crystal structure of rhodopsin in complex with a mini-Go sheds light on the principles of G protein selectivity. Sci Adv 4: eaat7052. doi: 10.1126/sciadv.aat7052
|
| [40] |
Lebon G, Bennett K, Jazayeri A, et al. (2011) Thermostabilisation of an agonist-bound conformation of the human adenosine A(2A) receptor. J Mol Biol 409: 298–310. doi: 10.1016/j.jmb.2011.03.075
|
| [41] |
Lebon G, Warne T, Edwards PC, et al. (2011) Agonist-bound adenosine A2A receptor structures reveal common features of GPCR activation. Nature 474: 521–525. doi: 10.1038/nature10136
|
| [42] |
Warne T, Serrano-Vega MJ, Baker JG, et al. (2008) Structure of a beta1-adrenergic G-protein-coupled receptor. Nature 454: 486–491. doi: 10.1038/nature07101
|
| [43] |
Lebon G, Edwards PC, Leslie AG, et al. (2015) Molecular determinants of CGS21680 binding to the human adenosine A2A receptor. Mol Pharmacol 87: 907–915. doi: 10.1124/mol.114.097360
|
| [44] |
Lebon G, Warne T, Tate CG (2012) Agonist-bound structures of G protein-coupled receptors. Curr Opin Struct Biol 22: 482–490. doi: 10.1016/j.sbi.2012.03.007
|
| [45] |
Xu F, Wu H, Katritch V, et al. (2011) Structure of an agonist-bound human A2A adenosine receptor. Science 332: 322–327. doi: 10.1126/science.1202793
|
| [46] |
Ye L, Van EN, Zimmer M, et al. (2016) Activation of the A2A adenosine G-protein-coupled receptor by conformational selection. Nature 533: 265–268. doi: 10.1038/nature17668
|
| [47] |
Strege A, Carpenter B, Edwards PC, et al. (2017) Strategy for the thermostabilization of an agonist-bound GPCR coupled to a G protein. Method Enzymol 594: 243–264. doi: 10.1016/bs.mie.2017.05.014
|
| [48] |
Carpenter B, Lebon G (2017) Human adenosine A2A receptor: Molecular mechanism of ligand binding and activation. Front Pharmacol 8: 898. doi: 10.3389/fphar.2017.00898
|
| [49] |
Liang YL, Khoshouei M, Radjainia M, et al. (2017) Phase-plate cryo-EM structure of a class B GPCR-G-protein complex. Nature 546: 118–123. doi: 10.1038/nature22327
|
| [50] |
Liang YL, Khoshouei M, Deganutti G, et al. (2018) Cryo-EM structure of the active, Gs-protein complexed, human CGRP receptor. Nature 561: 492–497. doi: 10.1038/s41586-018-0535-y
|
| [51] |
Zhang Y, Sun B, Feng D, et al. (2017) Cryo-EM structure of the activated GLP-1 receptor in complex with a G protein. Nature 546: 248–253. doi: 10.1038/nature22394
|
| [52] |
Koehl A, Hu H, Maeda S, et al. (2018) Structure of the micro-opioid receptor-Gi protein complex. Nature 558: 547–552. doi: 10.1038/s41586-018-0219-7
|
| [53] |
Draper-Joyce CJ, Khoshouei M, Thal DM, et al. (2018) Structure of the adenosine-bound human adenosine A1 receptor-Gi complex. Nature 558: 559–563. doi: 10.1038/s41586-018-0236-6
|
| [54] |
Kang Y, Kuybeda O, de Waal PW, et al. (2018) Cryo-EM structure of human rhodopsin bound to an inhibitory G protein. Nature 558: 553–558. doi: 10.1038/s41586-018-0215-y
|
| [55] | Khoshouei M, Radjainia M, Baumeister W, et al. (2017) Cryo-EM structure of haemoglobin at 3.2 A determined with the Volta phase plate. Nat Commun 8: 16099. |
| [56] |
Renaud JP, Chari A, Ciferri C, et al. (2018) Cryo-EM in drug discovery: Achievements, limitations and prospects. Nat Rev Drug Discov 17: 471–492. doi: 10.1038/nrd.2018.77
|
| [57] | Green SA, Holt BD, Liggett SB (1992) Beta 1- and beta 2-adrenergic receptors display subtype-selective coupling to Gs. Mol Pharmacol 41: 889–893. |
| [58] |
Niesen FH, Berglund H, Vedadi M (2007) The use of differential scanning fluorimetry to detect ligand interactions that promote protein stability. Nat Protoc 2: 2212–2221. doi: 10.1038/nprot.2007.321
|
| [59] |
Yen HY, Hoi KK, Liko I, et al. (2018) PtdIns(4,5)P2 stabilizes active states of GPCRs and enhances selectivity of G-protein coupling. Nature 559: 423–427. doi: 10.1038/s41586-018-0325-6
|
| [60] |
Gales C, Rebois RV, Hogue M, et al. (2005) Real-time monitoring of receptor and G-protein interactions in living cells. Nat Methods 2: 177–184. doi: 10.1038/nmeth743
|
| [61] |
Hein P, Frank M, Hoffmann C, et al. (2005) Dynamics of receptor/G protein coupling in living cells. EMBO J 24: 4106–4114. doi: 10.1038/sj.emboj.7600870
|
| [62] |
Dixon AS, Schwinn MK, Hall MP, et al. (2016) NanoLuc complementation reporter optimized for accurate measurement of protein interactions in cells. ACS Chem Biol 11: 400–408. doi: 10.1021/acschembio.5b00753
|
| [63] |
Irannejad R, Pessino V, Mika D, et al. (2017) Functional selectivity of GPCR-directed drug action through location bias. Nat Chem Biol 13: 799–806. doi: 10.1038/nchembio.2389
|
| [64] |
Irannejad R, Tomshine JC, Tomshine JR, et al. (2013) Conformational biosensors reveal GPCR signalling from endosomes. Nature 495: 534–538. doi: 10.1038/nature12000
|
| [65] | Kenakin T, Christopoulos A (2013) Signalling bias in new drug discovery: Detection, quantification and therapeutic impact. Nat Rev Drug Discov 12: 205–216. |
| [66] |
Gentry PR, Sexton PM, Christopoulos A (2015) Novel allosteric modulators of G protein-coupled receptors. J Biol Chem 290: 19478–19488. doi: 10.1074/jbc.R115.662759
|
| [67] |
Skiba NP, Bae H, Hamm HE (1996) Mapping of effector binding sites of transducin alpha-subunit using G alpha t/G alpha i1 chimeras. J Biol Chem 271: 413–424. doi: 10.1074/jbc.271.1.413
|
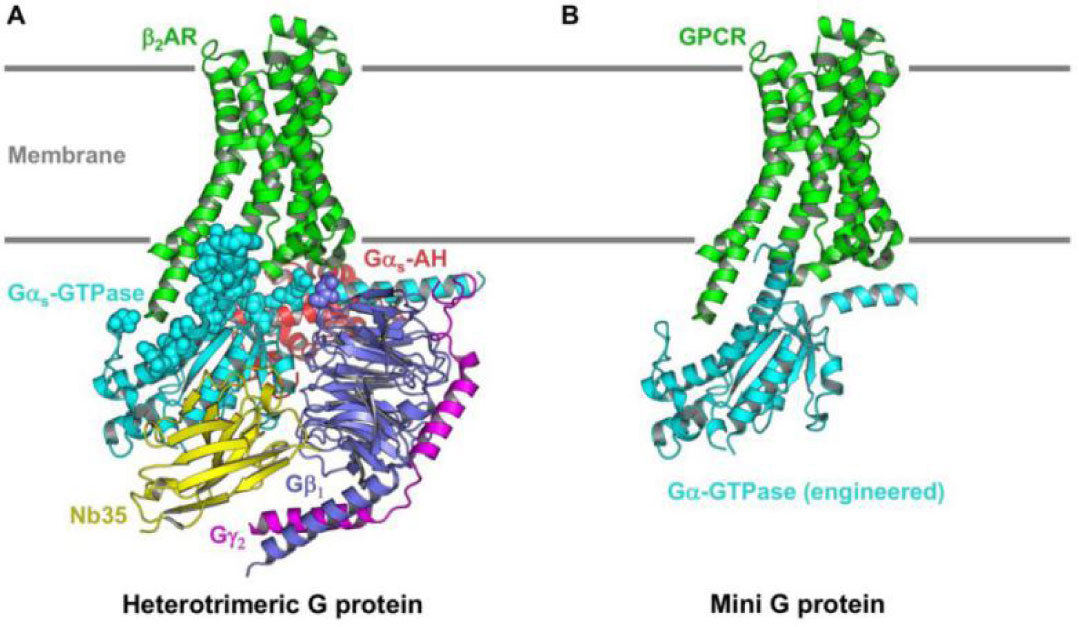
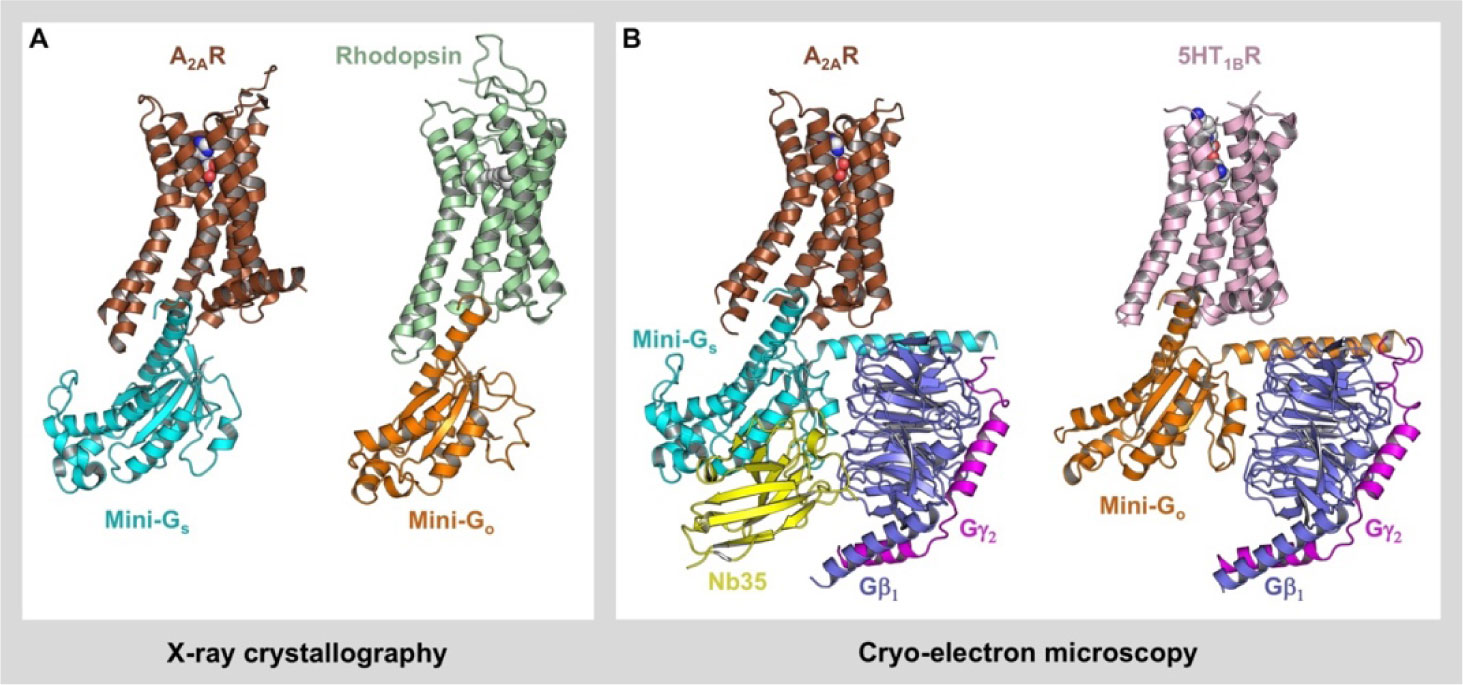
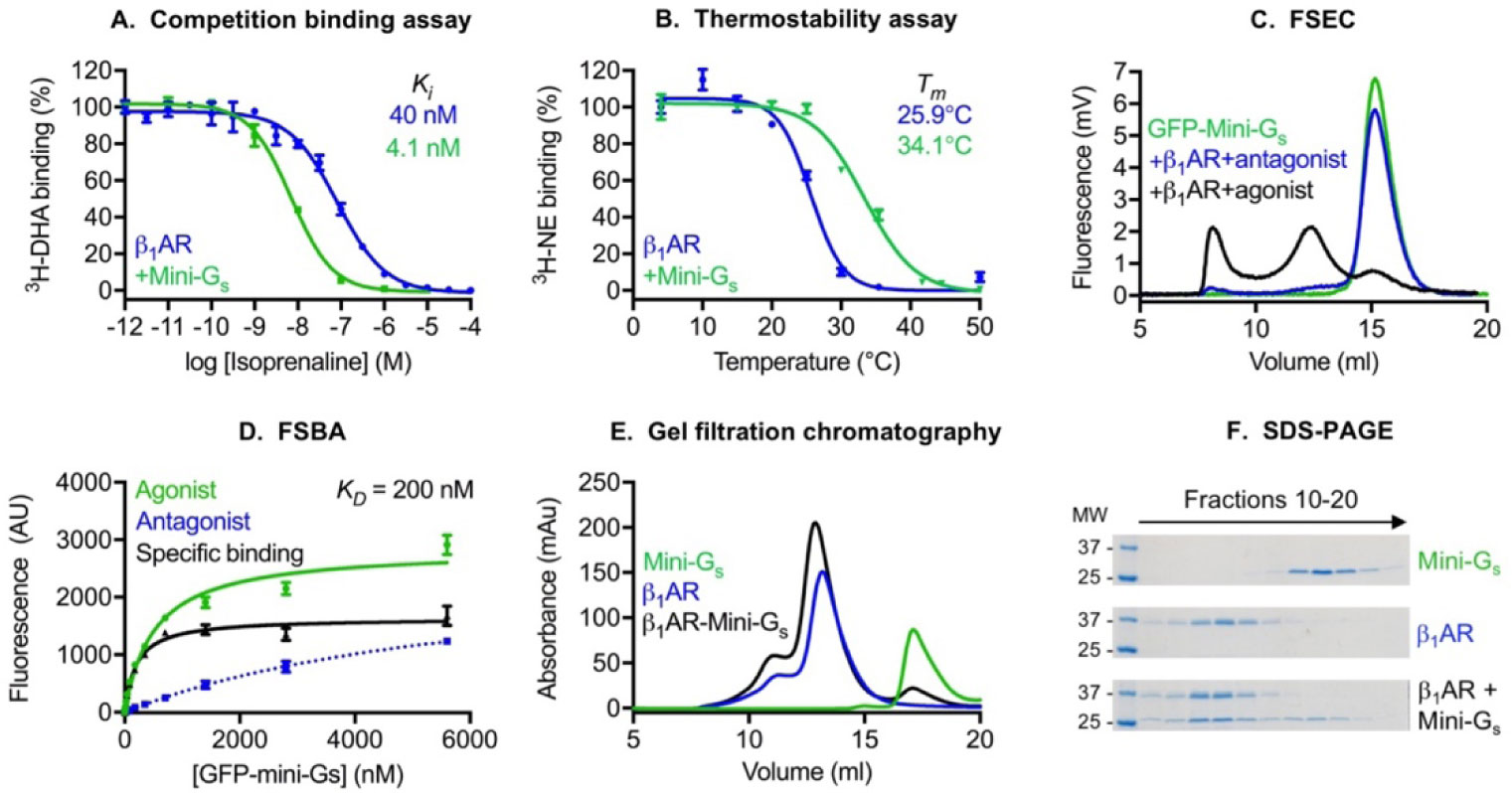
Figures(3) / Tables(1)

Byron Carpenter. Current applications of mini G proteins to study the structure and function of G protein-coupled receptors[J]. AIMS Bioengineering, 2018, 5(4): 209-225. doi: 10.3934/bioeng.2018.4.209






 DownLoad:
DownLoad: