Citation: Peter Romeo Nyarko, Martin Anokye. Mathematical modeling and numerical simulation of a multiscale cancer invasion of host tissue[J]. AIMS Mathematics, 2020, 5(4): 3111-3124. doi: 10.3934/math.2020200

| [1] | H. Jan, R. Kathy, W. Wendy, et al. A guide to cancer drug development and regulation, Astra Zeneca, 2006. |
| [2] | F. Cornelis, O. Saut, P. Comsille, et al. In vivo mathematical modeling of tumor growth from imaging data: Soon to come in the future? Diagn. Interv. Imag., 94 (2013), 593-600. |
| [3] |
G. Meral, C. Stinner and C. Surulescu, On a multiscale model involving cell contractivity and its effects on tumor invasion, Discrete and continuous dynamical systems series B, 20 (2014), 189-213. doi: 10.3934/dcdsb.2015.20.189

|
| [4] |
J. Kelkel and C. Surulescu, On some models for cancer cell migration through tissue networks, Math. Biosci. Eng., 8 (2011), 575-589. doi: 10.3934/mbe.2011.8.575
|
| [5] |
B. Yue, Biology of the Extracellular Matrix: An Overview, J. Glaucoma, 23 (2014), 20-23. doi: 10.1097/IJG.0000000000000108
|
| [6] |
M. A. J. Chaplain, G. Lolas, Mathematical modelling of cancer invasion of tissue: Dynamic heterogeneity, Netw. Heterog. Media, 1 (2006), 399-439. doi: 10.3934/nhm.2006.1.399
|
| [7] | N. Kolbem, A tumor invasion model for heterogeneous cancer cell populations: mathematical analysis and numerical methods, Ph.D thesis, der Johannes Gutenberg-Universität Mainz, 2017. Available from: https://https://publications.ub.uni-mainz.de/theses/volltexte/2018/100001842/pdf/100001842.pdf. |
| [8] |
N. Sfakianakis, N. Kolbe, N. Hellmann, et al. 2016. A multiscale approach to the migration of cancer stem cells: mathematical modelling and simulations, B. Math. Biol., 79 (2017), 209-235. doi: 10.1007/s11538-016-0233-6
|
| [9] |
P. Domschke, D. Trucu, A. Gerisch, et al. Mathematical modelling of cancer invasion: Implications of cell adhesion variability for tumor infinitive growth patterns, J. Theor. Biol., 361 (2014), 41-60. doi: 10.1016/j.jtbi.2014.07.010
|
| [10] |
V. Andasari, A. Gerisch, G. Lolas, et al. Mathematical modelling of cancer cell invasion of tissue: biological insight from mathematical analysis and computational simulation, J. Math. Biol., 63 (2011), 141-171. doi: 10.1007/s00285-010-0369-1
|
| [11] |
P. Lu, T. Dumitru, L. Ping, et al. Multiscale Mathematical Model of Tumour Invasive Growth, B. Math. Biol., 79 (2017), 389-429. doi: 10.1007/s11538-016-0237-2
|
| [12] | T. Dumitru, Multiscale Modelling of Cancer Invasion, 6th European Conference on Computational Mechanics (ECCM 6), 2018. |
| [13] |
R. M. Sutherland, Cell and environment interaction in tumour microregions: the multicell spheroid model, Science, 240 (1988), 177-184. doi: 10.1126/science.2451290
|
| [14] | N. Kolbe, M. Lukacova-Medvid ova, N. Sfakianakis, et al. Numerical simulation of a contractivity based multiscale cancer invasion model, Institute of Math., Johannes Gutenberg-Uni., Mainz, Germany, 2016. |
| [15] |
J. A. Sherratt, M. J. A. Chaplain, A new mathematical model for avascular tumour growth, J. Math. Biol., 43 (2001), 291-312. doi: 10.1007/s002850100088
|
| [16] |
Y. Tao, C. Cui, A density-dependent chemotaxis-haptotaxis system modeling cancer invasion, J. Math. Anal. Appl., 367 (2010), 612-624. doi: 10.1016/j.jmaa.2010.02.015
|
| [17] | R. A. Gatenby, E. T. Gawlinski, A reaction-diffusion model of cancer invasion, Cancer Res., 56 (1996), 5745-5753. |
| [18] |
P. Domschke, D. Trucu, A. Gerisch, et al. Mathematical modelling of cancer invasion: Implications of cell adhesion variability for tumour infiltrative growth patterns, J. Theor. Biol., 361 (2014), 40-61. doi: 10.1016/j.jtbi.2014.07.010
|
| [19] | A. Zhigun, C. Surulescu, A. Hunt, Global existence for a degenerate haptotaxis model of tumor invasion under the go-or-grow dichotomy hypothesis, preprint. |
| [20] |
W. Mueller-Klieser, Method for determination of oxygen consumption rates and diffusion coefficients in multicellular spheroids, Biophys. J., 46 (1984), 343-348. doi: 10.1016/S0006-3495(84)84030-8
|
| [21] |
W. Mueller-Klieser, J. P. Freyer, R. M. Sutherland, Influence of glucose and oxygen supply conditions on the oxygenation of multicellular spheroids, Brit. J. Cancer, 53 (1986), 345-353. doi: 10.1038/bjc.1986.58
|
| [22] |
T. Roose, S. J. Chapman, P. K. Maini, Mathematical Models of Avascular Tumor Growth, Siam rev., 49 (2007), 179-208. doi: 10.1137/S0036144504446291
|
| [23] | T. S. Deisboeck, Z. Wang, P. Macklin, et al. Multiscale Cancer Modeling, Annual review of biomedical engineering, 2010. |
| [24] |
D. Hanahan and R. A. Weinberg, The Hallmarks of Cancer, Cell, 100 (2000), 57-70. doi: 10.1016/S0092-8674(00)81683-9
|
| [25] |
D. Hanahan, R. A. Weinberg, Hallmarks of cancer: the next generation, Cell, 144 (2011), 646-674. doi: 10.1016/j.cell.2011.02.013
|
| [26] |
B. Qian, J. W. Pollard, Macrophage Diversity Enhances Tumor Progression and Metastasis, Cell, 141 (2010), 39-51. doi: 10.1016/j.cell.2010.03.014
|
| [27] |
J. A. Joyce, J. W. Pollard, Microenvironmental regulation of metastasis, Nat. Rev. Cancer, 9 (2009), 239-252. doi: 10.1038/nrc2618
|
| [28] |
M. A. J. Chaplain and G. Lolas, Mathematical modelling of cancer cell invasion of tissue: The role of the urokinase plasminogen activation system, Math. Mod. Meth. Appl. S., 15 (2005), 1685-1734. doi: 10.1142/S0218202505000947
|
| [29] |
V. Andasari, R. T. Roper, M. H. Swat, et al. Integrating Intracellular Dynamics Using CompuCell3D and Bionetsolver: Applications to Multiscale Modelling of Cancer Cell Growth and Invasion, PLoS ONE, 7 (2012), e33726. doi: 10.1371/journal.pone.0033726
|
| [30] | M. A. J. Chaplain and N. Deakin, Mathematical modeling of cancer invasion: the role of membrane-bound matrix metalloproteinases, Frontiers in oncology, 2013. |
| [31] | C. Walker, E. Mojares, A. D. R. Hernández, Role of Extracellular Matrix in Development and Cancer Progression, Int. J. Mol. Sci., 19 (2018), 3028. |
| [32] | Y. T. N. Edalgo, A. N. F. Versypt, Mathematical Modeling of Metastatic Cancer Migration through a Remodeling Extracellular Matrix, Processes, 6 (2018), 58. |
| [33] |
P. J. Murray, C. M. Edwards, M. J. Tindall, et al. From a discrete to a continuum model of cell dynamics in one dimension, Phys. Rev. E, 80 (2009), 31912. doi: 10.1103/PhysRevE.80.031912
|
| [34] |
A. Gerischa, M. A. J. Chaplain, Robust numerical methods for taxis-diffusion-reaction systems: Applications to biomedical problems, Math. Comput. Model., 43 (2006), 49-75. doi: 10.1016/j.mcm.2004.05.016
|
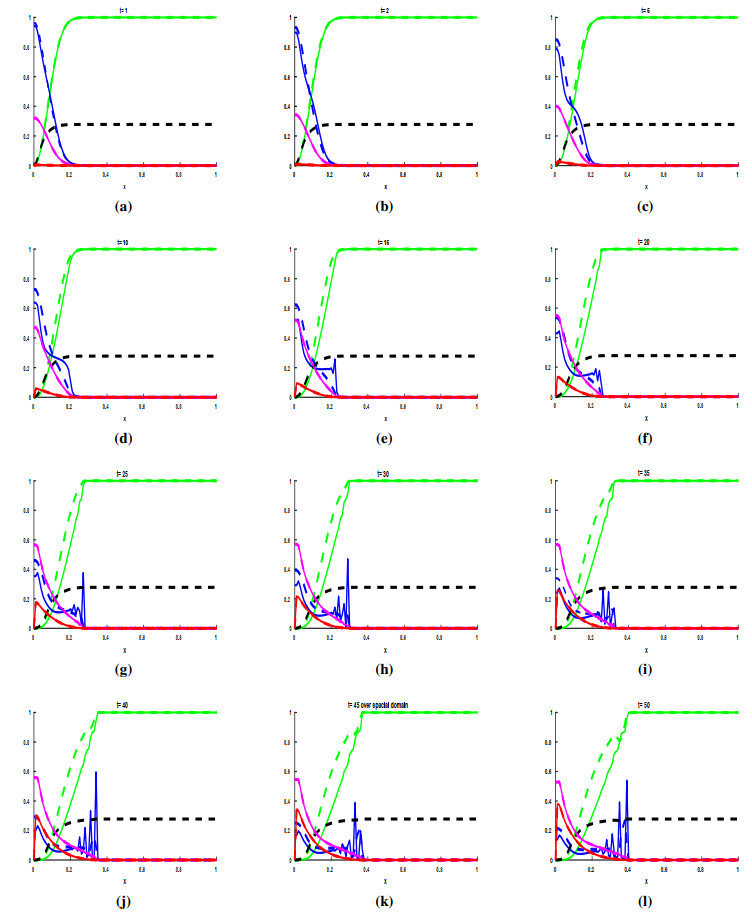
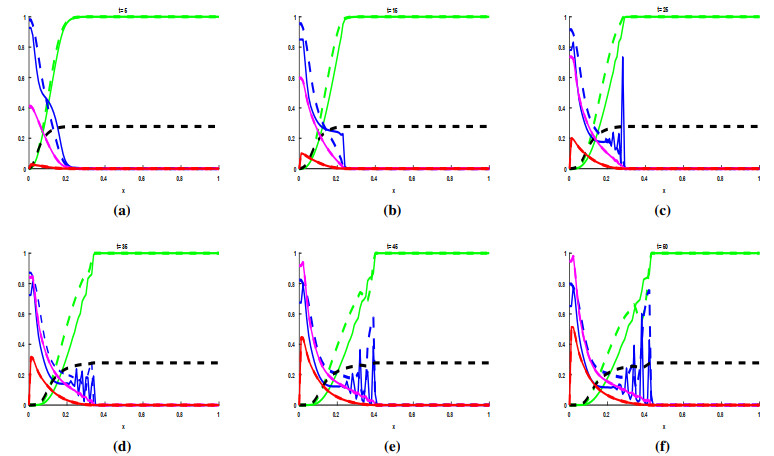
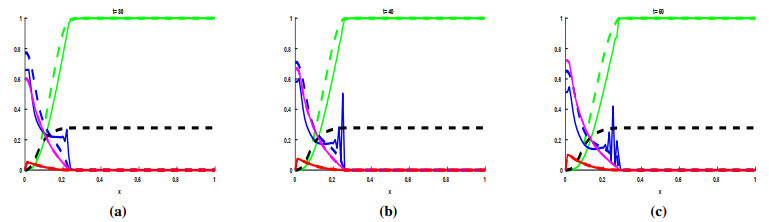
Figures(3) / Tables(1)

Peter Romeo Nyarko, Martin Anokye. Mathematical modeling and numerical simulation of a multiscale cancer invasion of host tissue[J]. AIMS Mathematics, 2020, 5(4): 3111-3124. doi: 10.3934/math.2020200







 DownLoad:
DownLoad: