Citation: Jianfeng Wen, Yanjin Liu, Yunjie Tu and Mark W. LeChevallier. Energy and chemical efficient nitrogen removal at a full-scale MBR water reuse facility[J]. AIMS Environmental Science, 2015, 2(1): 42-55. doi: 10.3934/environsci.2015.1.42

| [1] |
Zhou H, Smith DW (2001) Advanced technologies in water and wastewater treatment. Can J Civil Eng 28: 49-66. doi: 10.1139/cjce-28-S1-49

|
| [2] |
Melin T, Jefferson B, Bixio D, et al. (2006) Membrane bioreactor technology for wastewater treatment and reuse. Desalination 187: 271-282. doi: 10.1016/j.desal.2005.04.086
|
| [3] | Verrecht B, Judd SJ, Guglielmi G, et al. (2008) An aeration energy model for an immersed membrane bioreactor. Water Res 42: 4716-4770. |
| [4] |
Schleper C, Nicol GW (2010) Ammonia-oxidising archaea - physiology, ecology and evolution. Adv Microb Physiol 57: 1-36. doi: 10.1016/B978-0-12-381045-8.00001-1
|
| [5] |
Giraldo E, Jjemba P, Liu Y, et al. (2011) Ammonia oxidizing archaea, AOA, population and kinetic changes in a full scale simultaneous nitrogen and phosphorus removal MBR. Proc Water Environ Federation 2011: 3156-3168. doi: 10.2175/193864711802721596
|
| [6] | Huang T, Li N, Huang Y (2012) Modelling of nitrogen removal and control strategy in continuous-flow-intermittent-aeration process. Afr J Biotechnol 11: 10626-10631. |
| [7] |
Guglielmi G, Andreottola G (2011) Alternate anoxic/aerobic operation for nitrogen removal in a membrane bioreactor for municipal wastewater treatment. Water Sci Techn 64: 1730-1735. doi: 10.2166/wst.2011.755
|
| [8] |
Curko J, Matosic M, Jakopovic HK, et al. (2010) Nitrogen removal in submerged MBR with intermittent aeration. Desalination Water Treat 24: 7-19. doi: 10.5004/dwt.2010.1118
|
| [9] |
Parikh C, Trivedi H, Livingston D (2011) A decade of simultaneous nitrification and denitrification experience in over 60 conventional and MBR applications - lessons learned. Proc Water Environ Federation 2011: 3629-3655. doi: 10.2175/193864711802721901
|
| [10] |
Holman JB, Wareham DG (2005) COD, ammonia and dissolved oxygen time profiles in the simultaneous nitrification/denitrification process. Biochem Eng J 22: 125-133. doi: 10.1016/j.bej.2004.09.001
|
| [11] |
Fu Z, Yang F, An Y, et al. (2009) Simultaneous nitrification and denitrification coupled with phosphorus removal in a modified anoxic/oxic-membrane bioreactor (A/O-MBR). Biochem Eng J 43: 191-196. doi: 10.1016/j.bej.2008.09.021
|
| [12] |
Li YZ, He YL, Ohandja DG, et al. (2008) Simutaneous nitrification-denitrification achieved by an improved internal-loop airlift MBR: comparative study. Bioresource Technol 99: 5867-5872. doi: 10.1016/j.biortech.2007.10.001
|
| [13] |
Sarioglu M, Insel G, Artan N, et al. (2009) Model evaluation of simultaneous nitrification and denitrification in a membrane bioreactor operated without an anoxic reactor. J Membrane Sci 337: 17-27. doi: 10.1016/j.memsci.2009.03.015
|
| [14] |
Wyffels S, Van Hulle SWH, Boeckx P, et al. (2004) Modelling and simulation of oxygen-limited partial nitrification in a membrane-assisted bioreactor (MBR). Biotechnol Bioeng 86: 531-542. doi: 10.1002/bit.20008
|
| [15] |
Daebel H, Manser R, Gujer W (2007) Exploring temporal variations of oxygen saturation constants of nitrifying bacteria. Water Res 41: 1094-1102. doi: 10.1016/j.watres.2006.11.011
|
| [16] |
Sharp R, Dailey S, Motyl M, et al. (2012) KDO experiments at NYC DEP's full-scale demonstration facility. Proc Water Environ Federation 2012: 4176-4182. doi: 10.2175/193864712811708482
|
| [17] |
Rieger L, Jones RM, Dold PL, et al. (2014) Ammonia-based feedforward and feeback aeration control in activated sludge process. Water Environ Res 86: 63-73. doi: 10.2175/106143013X13596524516987
|
| [18] | Henze M, Gujer W, Mino T, et al. (2000). Activated sludge models: ASM1, ASM2, ASM2d and ASM3. IWA Publishing, London. |
| [19] | Tchobanoglous G, Burton FL, Stensel HD (2004) Wastewater engineering, treatment and reuse, 4 Eds., Singapore: McGraw-Hill Education, 690. |
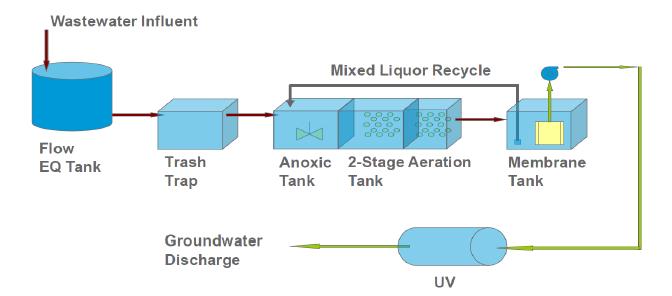

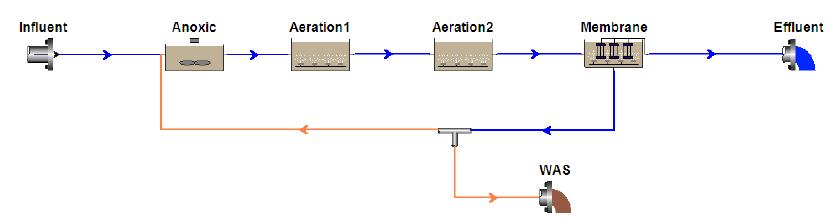
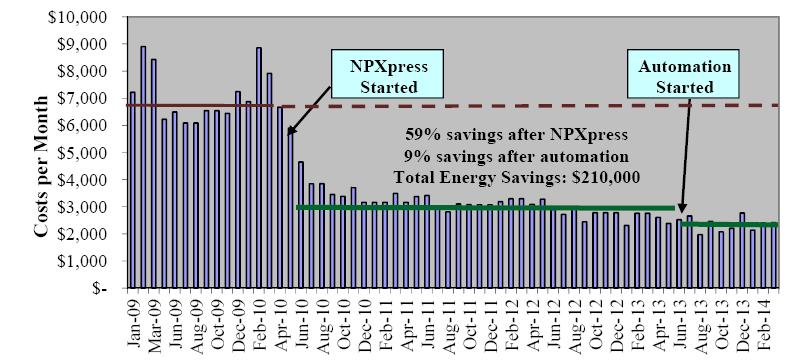
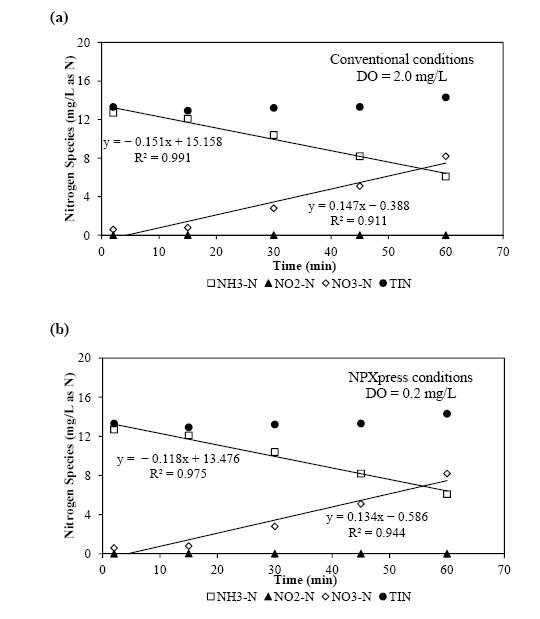

Figures(10) / Tables(1)

Jianfeng Wen, Yanjin Liu, Yunjie Tu and Mark W. LeChevallier. Energy and chemical efficient nitrogen removal at a full-scale MBR water reuse facility[J]. AIMS Environmental Science, 2015, 2(1): 42-55. doi: 10.3934/environsci.2015.1.42







 DownLoad:
DownLoad: