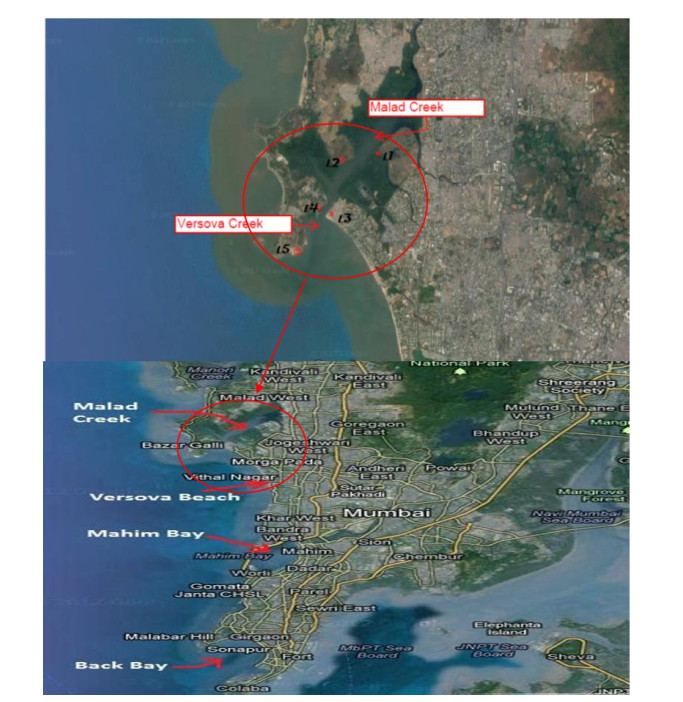
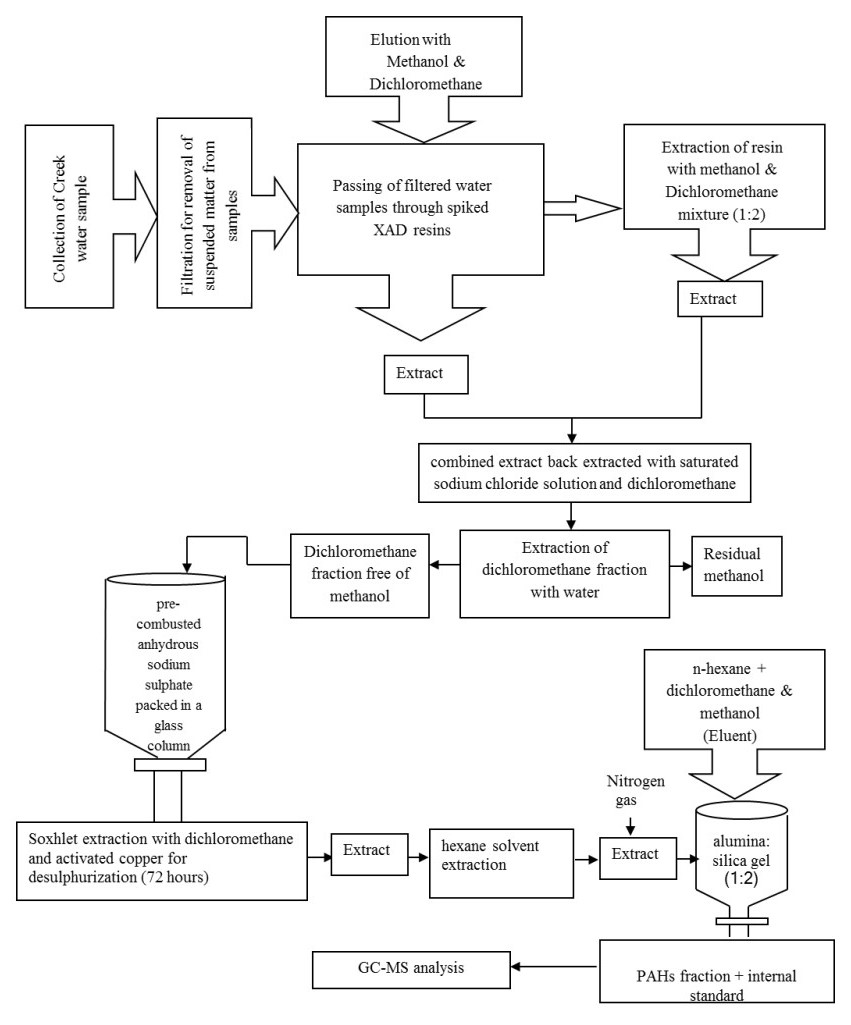
The carcinogenic and endocrine-disrupting PAHs were investigated in surface water of the north-western creeks of India. The concentrations of ΣPAHs were found to vary in the range of 114.32-347.04 μg L-1, (mean 224.78 ± 8.85 μg L-1), out of which 49.12% contribution is due to ΣC-PAHs. The assessment of toxicity and biological risk arising due to individual C-PAHs was made by calculating their toxic equivalent quantity. The level of individual C-PAHs was reported exceeding the final chronic values, Canadian water quality guideline values and Netherlands maximum permissible concentration values set for the protection of aquatic life. The mean BaP concentration (10.32 ± 2.75 μg L-1) was above the European Directive 2008/105/EC Environmental Quality Standards (EQS) value; while the sum of BkF + BbF (26.76 μg L-1) and BghiP + InP (19.59 μg L-1) were significantly higher than that set by the EQS. The results of the present study will help in understanding the global distribution and fate of PAHs which is required for implementing the necessary steps towards mitigation of the ecotoxicological risk arising due to the existence of such contaminants in the aquatic environment across the world.
Citation: P. U. Singare, J.P. Shirodkar. Persistent and carcinogenic polycyclic aromatic hydrocarbons in the north-western coastal marine environment of India[J]. AIMS Environmental Science, 2021, 8(2): 169-189. doi: 10.3934/environsci.2021012
The carcinogenic and endocrine-disrupting PAHs were investigated in surface water of the north-western creeks of India. The concentrations of ΣPAHs were found to vary in the range of 114.32-347.04 μg L-1, (mean 224.78 ± 8.85 μg L-1), out of which 49.12% contribution is due to ΣC-PAHs. The assessment of toxicity and biological risk arising due to individual C-PAHs was made by calculating their toxic equivalent quantity. The level of individual C-PAHs was reported exceeding the final chronic values, Canadian water quality guideline values and Netherlands maximum permissible concentration values set for the protection of aquatic life. The mean BaP concentration (10.32 ± 2.75 μg L-1) was above the European Directive 2008/105/EC Environmental Quality Standards (EQS) value; while the sum of BkF + BbF (26.76 μg L-1) and BghiP + InP (19.59 μg L-1) were significantly higher than that set by the EQS. The results of the present study will help in understanding the global distribution and fate of PAHs which is required for implementing the necessary steps towards mitigation of the ecotoxicological risk arising due to the existence of such contaminants in the aquatic environment across the world.

| [1] |
Kanzari F, Syakti AD, Asia L, et al. (2014) Distributions and sources of persistent organic pollutants (aliphatic hydrocarbons, PAHs, PCBs and pesticides) in surface sediments of an industrialized urban river (Huveaune), France. Sci Total Environ 478: 141-151. doi: 10.1016/j.scitotenv.2014.01.065

|
| [2] |
Abdel-Shafy HI, Mansour MSM (2016) A review on polycyclic aromatic hydrocarbons: source, environmental impact, effect on human health and remediation. Egypt J Pet 25: 107-123. doi: 10.1016/j.ejpe.2015.03.011
|
| [3] |
Lin Y, Qiu X, Ma Y, et al. (2015) Concentrations and spatial distribution of polycyclic aromatic hydrocarbons (PAHs) and nitrated PAHs (NPAHs) in the atmosphere of North China, and the transformation from PAHs to NPAHs. Environ Pollut 196: 164-170. doi: 10.1016/j.envpol.2014.10.005
|
| [4] |
Friedman CL, Pierce JR, Selin NE (2014) Assessing the Influence of Secondary Organic versus Primary Carbonaceous Aerosols on Long-Range Atmospheric Polycyclic Aromatic Hydrocarbon Transport. Environ Sci Technol 48: 3293-3302. doi: 10.1021/es405219r
|
| [5] |
Basavaiah N, Mohite RD, Singare PU, et al. (2017) Vertical distribution, composition profiles, sources and toxicity assessment of PAH residues in the reclaimed mudflat sediments from the adjacent Thane Creek of Mumbai. Mar Pollut Bull 118: 112-124. doi: 10.1016/j.marpolbul.2017.02.049
|
| [6] | Singare PU (2015) Studies on Polycyclic Aromatic Hydrocarbons in Sediments of Mithi River of Mumbai, India: Assessment of Sources, Toxicity Risk and Biological Impact. Mar Pollut Bull 101: 232-242. |
| [7] |
Tongo I, Ezemonye L, Akpeh K (2017) Levels, distribution and characterization of polycyclic aromatic hydrocarbons (PAHs) in Ovia river, Southern Nigeria. J Environ Chem Eng 5: 504-512. doi: 10.1016/j.jece.2016.12.035
|
| [8] |
Hong WJ, Jia H, Li YF et al. (2016) Polycyclic aromatic hydrocarbons (PAHs) and alkylated PAHs in the coastal seawater, surface sediment and oyster from Dalian, Northeast China. Ecotoxicol Environ Saf 128: 11-20. doi: 10.1016/j.ecoenv.2016.02.003
|
| [9] | Qin XB, Sun HW, Wang CP, et al. (2010) Impacts of crab bioturbation on the fate of polycyclic aromatic hydrocarbons in sediment from the Beitang Estuary of Tianjin, China. Environ Toxicol Chem 29: 1248-1255. |
| [10] |
Gu YG, Lin Q, Lu TT, et al. (2013) Levels, composition profiles and sources of polycyclic aromatic hydrocarbons in surface sediments from Nan'ao Island, a representative mariculture base in South China. Mar Pollut Bull 75: 310-316. doi: 10.1016/j.marpolbul.2013.07.039
|
| [11] |
Hawliczek A, Nota B, Cenijn P, et al. (2012) Developmental toxicity and endocrine disrupting potency of 4-azapyrene, benzo[b]fluorene and retene in the zebrafish Danio rerio. Reprod Toxicol 33: 213-223. doi: 10.1016/j.reprotox.2011.11.001
|
| [12] |
Liu LY, Wang JZ, Wei GL, et al. (2012) Polycyclic aromatic hydrocarbons (PAHs) in continental shelf sediment of China: implications for anthropogenic influences on coastal marine environment. Environ Pollut 167: 155-162. doi: 10.1016/j.envpol.2012.03.038
|
| [13] |
Lewis MA, Russel MJ (2015) Contaminant profiles for surface water, sediment, flora and fauna associated with the mangrove fringe along middle and lower eastern Tampa Bay. Mar Pollut Bull 95: 273-282. doi: 10.1016/j.marpolbul.2015.04.001
|
| [14] |
Santana JL, Massone CG, Valdes M, et al. (2015) Occurrence and source appraisal of polycyclic aromatic hydrocarbons (PAHs) in surface waters of the Almendares River, Cuba. Arch Environ Contam Toxicol 69: 143-152. doi: 10.1007/s00244-015-0136-9
|
| [15] |
Sarria-Villa R, Ocampo-Duque W, Paez M, et al. (2016) Presence of PAHs in water and sediments of the Colombian Cauca River during heavy rain episodes, and implications for risk assessment. Sci Total Environ 540: 455-465. doi: 10.1016/j.scitotenv.2015.07.020
|
| [16] |
Singare PU (2016) Carcinogenic and endocrine disrupting PAHs in the aquatic ecosystem of India. Environ Monit Assess 188: 1-25. doi: 10.1007/s10661-015-4999-z
|
| [17] |
Yan J, Liu J, Shi X, et al. (2016) Polycyclic aromatic hydrocarbons (PAHs) in water from three estuaries of China: distribution, seasonal variations and ecological risk assessment. Mar Pollut Bull 109: 471-479. doi: 10.1016/j.marpolbul.2016.05.025
|
| [18] |
Manoli E, Samara C, Konstantinou I, et al. (2000) Polycyclic aromatic hydrocarbons in the bulk precipitation and surface waters of Northern Greece. Chemosphere 41: 1845-1855. doi: 10.1016/S0045-6535(00)00134-X
|
| [19] | Mane S, Sundaram S (2014) Studies on some aspects on the biology of green mussel Perna viridis (Linnaeus, 1758) from Versova creek, Mumbai, northwest coast of India. Int Res J Sci Eng 2: 47-50. |
| [20] |
Shirke S, Pinto SM, Kushwaha VK, et al. (2016) Object-based image analysis for the impact of sewage pollution in Malad Creek, Mumbai, India. Environ Monit Assess 188: 95-99. doi: 10.1007/s10661-015-4981-9
|
| [21] |
Zeng EY, Yu CC, Tran K (1999) In situ measurements of chlorinated hydrocarbons in the water column off the Palos Verdes Peninsula, California. Environ Sci Technol 33: 392-398. doi: 10.1021/es980561e
|
| [22] | WHO (1998) Polynuclear aromatic hydrocarbons. Guidelines for Drinking-Water Quality, 2nd edition. Addendum to Vol. 2 Health Criteria and Other Supporting Information. World Health Organization, Geneva, pp. 123-152. |
| [23] |
Xiang N, Jiang C, Yang T, et al. (2018) Occurrence and distribution of Polycyclic aromatic hydrocarbons (PAHs) in seawater, sediments and corals from Hainan Island, China. Ecotoxicol Environ Saf 152: 8-15. doi: 10.1016/j.ecoenv.2018.01.006
|
| [24] |
Santos E, Souza MRR, Vilela Junior AR, et al. (2018) Polycyclic aromatic hydrocarbons (PAH) in superficial water from a tropical estuarine system: Distribution, seasonal variations, sources and ecological risk assessment. Mar Pollut Bull 127: 352-358. doi: 10.1016/j.marpolbul.2017.12.014
|
| [25] | Nwineewii JD, Marcus AC (2015) Polycyclic Aromatic Hydrocarbons (PAHs) In Surface Water and Their Toxicological Effects in Some Creeks of South East Rivers State (Niger Delta) Nigeria. IOSR J Environ Sci Toxicol Food Technol 9: 27-30. |
| [26] |
Yang D, Qi SH, Zhang Y, et al. (2013) Levels, sources and potential risks of polycyclic aromatic hydrocarbons (PAHs) in multimedia environment along the Jinjiang River mainstream to Quanzhou Bay, China. Mar Pollut Bull 76: 298-306. doi: 10.1016/j.marpolbul.2013.08.016
|
| [27] |
Adeniji AO, Okoh OO, Okoh AI (2019) Levels of Polycyclic Aromatic Hydrocarbons in the Water and Sediment of Bufalo River Estuary, South Africa and Their Health Risk Assessment. Arch Environ Contam Toxicol 76: 657-669. doi: 10.1007/s00244-019-00617-w
|
| [28] |
Edokpayi JN, Odiyo JO, Popoola OE, et al. (2016) Determination and Distribution of Polycyclic Aromatic Hydrocarbons in Rivers, Sediments and Wastewater Effluents in Vhembe District, South Africa. Int J Environ Res Public Health 13: 387, 1-12. doi: 10.3390/ijerph13040387
|
| [29] | Nekhavhambe TJ., van Ree T, Fatoki OS (2014) Determination and distribution of polycyclic aromatic hydrocarbons in rivers, surface runoff, and sediments in and around Thohoyandou, Limpopo Province, South Africa. Water SA 40: 415-424. |
| [30] | Eganhouse RP, Simoneit BRT, Kaplan IR (1981) Extractable organic matter in urban stormwater runoff. 2. Molecular characterization. Environ Sci Technol 15: 315-326. |
| [31] |
Hoffman EJ, Mills GL, Latimer JS, et al. (1984) Urban runoff as a source of polycyclic aromatic hydrocarbons to coastal waters. Environ Sci Technol 18: 580-587. doi: 10.1021/es00126a003
|
| [32] |
Agarwal T, Khillare P, Shridhar V, et al. (2009) Pattern, sources and toxic potential of PAHs in the agricultural soils of Delhi, India. J Hazard Mater 163: 1033-1039. doi: 10.1016/j.jhazmat.2008.07.058
|
| [33] |
Xing XL, Qi S, Zhang J, et al. (2011) Spatial distribution and source diagnosis of polycyclic aromatic hydrocarbons in soils from Chengdu Economic Region, Sichuan Province, western China. J Geochem Explor 110: 146-154. doi: 10.1016/j.gexplo.2011.05.001
|
| [34] |
Sprovieri M, Feo ML, Prevedello L, et al. (2007) Heavy metals, polycyclic aromatic hydrocarbons and polychlorinated biphenyls in surface sediments of the Naples harbor (southern Italy). Chemosphere 67: 998-1009. doi: 10.1016/j.chemosphere.2006.10.055
|
| [35] |
Ravindra K, Wauters E, Grieken RV (2008) Variation in particulate PAHs levels and their relation with the transboundary movement of the air masses. Sci Total Environ 396: 100-110. doi: 10.1016/j.scitotenv.2008.02.018
|
| [36] |
Tobiszewski M, Namiesnik J (2012) PAH diagnostic ratios for the identification of pollution emission sources. Environ Pollut 162: 110-119. doi: 10.1016/j.envpol.2011.10.025
|
| [37] |
Cao ZH, Wang YQ, Ma YM, et al. (2005) Occurrence and distribution of polycyclic aromatic hydrocarbons in reclaimed water and surface water of Tianjin, China. J Hazard Mater 122: 51-59. doi: 10.1016/j.jhazmat.2005.04.003
|
| [38] |
Boonyatumanond R, Wattayakorn G, Togo A, et al. (2006) Distribution and origins of polycyclic aromatic hydrocarbons (PAHs) in riverine, estuarine, and marine sediments in Thailand. Mar Pollut Bull52: 942-956. doi: 10.1016/j.marpolbul.2005.12.015
|
| [39] | Mostert MMR., Ayoko GA, Kokot S (2010) Application of chemometrics to analysis of soil pollutants. Trends Anal Chem 29: 430-435. |
| [40] |
Mai BX, Qi SH, Zeng EY, et al. (2003) Distribution of polycyclic aromatic hydrocarbons in the coastal region off Macao, China: assessment of input sources and transport pathways using compositional analysis. Environ Sci Technol 37: 4855-4863. doi: 10.1021/es034514k
|
| [41] |
Rocher V, Azimi S, Moilleron R, et al. (2004) Hydrocarbons and heavy metals in the different sewer deposits in the Le Marais' catchment (Paris, France): stocks, distributions and origins. Sci Total Environ 323: 107-122. doi: 10.1016/j.scitotenv.2003.10.010
|
| [42] |
Wang XC, Sun S, Ma HQ, et al. (2006) Sources and distribution of aliphatic and polyaromatic hydrocarbons in sediments of Jiaozhou Bay, Qingdao, China. Mar Pollut Bull 52: 129-138. doi: 10.1016/j.marpolbul.2005.08.010
|
| [43] |
Montuori P, Aurino S, Garzonio F, et al. (2016) Distribution, sources and ecological risk assessment of polycyclic aromatic hydrocarbons in water and sediments from Tiber River and estuary, Italy. Sci Total Environ 566-567: 1254-1267. doi: 10.1016/j.scitotenv.2016.05.183
|
| [44] |
Zhang W, Zhang S, Wan C, et al. (2008) Source diagnostics of polycyclic aromatic hydrocarbons in urban road runoff, dust, rain and canopy throughfall. Environ Pollut 153: 594-601. doi: 10.1016/j.envpol.2007.09.004
|
| [45] |
Chung MK, Hu R, Cheung KC, et al. (2007) Pollutants in Hongkong soils: polycyclicaromatic hydrocarbons. Chemosphere 67: 464-473. doi: 10.1016/j.chemosphere.2006.09.062
|
| [46] |
Li G, Xia X, Yang Z, et al. (2006) Distribution and sources of polycyclic aromatic hydrocarbons in the middle and lower reaches of the Yellow River, China. Environ Pollut 144: 985-993. doi: 10.1016/j.envpol.2006.01.047
|
| [47] |
De La Torre-Roche RJ, Lee WY, Campos-Diaz SI (2009) Soil-borne polycyclic aromatic hydrocarbons in El Paso, Texas: analysis of a potential problem in the United States/Mexico border region. J Hazard Mater 163: 946-958. doi: 10.1016/j.jhazmat.2008.07.089
|
| [48] |
Akyuz M, Cabuk H (2010) Gaseparticle partitioning and seasonal variation of polycyclic aromatic hydrocarbons in the atmosphere of Zonguldak, Turkey. Sci Total Environ 408: 5550-5558. doi: 10.1016/j.scitotenv.2010.07.063
|
| [49] | Dhananjayan V, Muralidharan S, Peter VR (2012) Occurrence and distribution of polycyclic aromatic hydrocarbons in water and sediment collected along the Harbour Line, Mumbai, India. Int J Oceanogr Article ID 403615, 7. |
| [50] |
Katsoyiannis A, Sweetman AJ, Jones KC (2011) PAH molecular diagnostic ratios applied to atmospheric sources: a critical evaluation using two Decades of source Inventory and air concentration data from the UK. Environ Sci Technol 45: 8897-8906. doi: 10.1021/es202277u
|
| [51] |
Pozo K, Perra G, Menchi V, et al. (2011) Levels and spatial distribution of polycyclic aromatic hydrocarbons (PAHs) in sediments from Lenga Estuary, central Chile. Mar Pollut Bull 62: 1572-1576. doi: 10.1016/j.marpolbul.2011.04.037
|
| [52] | Law RJ, Dawes VJ, Woodhead RJ, et al. (1997) Polycyclic aromatic hydrocarbons (PAH) in seawater around England and Wales. Mar Pollut Bull 34: 306-322. |
| [53] |
Barron MG, Podrabsky T, Ogle S, et al. (1999) Are aromatic hydrocarbons the primary determinant of petroleum toxicity to aquatic organisms? Aquat Toxicol 46: 253-268. doi: 10.1016/S0166-445X(98)00127-1
|
| [54] | Agroudy NA, Soliman YA, Hamed MA, et al. (2017) Distribution of PAHs in Water, Sediments Samples of Suez Canal During 2011. J Aquat Pol Toxicol 1: 1-10. |
| [55] | Pohl A, Kostecki M, Jureczko I, et al. (2018) Polycyclic aromatic hydrocarbons in water and bottom sediments of a shallow, lowland dammed reservoir (on the example of the reservoir Blachownia, South Poland). Arch Environ Prot 44: 10-23. |
| [56] | US Environmental Protection Agency (USEPA), (2012) Regional screening levels for chemical contaminants at superfund sites. Regional screening table. User's guide. (Access date: November 2012). < http://www.epa.gov/reg3hwmd/risk/human/rb-concentration_table/usersguide.htm > . |
| [57] | Di Toro DM, McGrath JA, Hansen DJ (2000) Technical basis for narcotic chemicals and polycyclic aromatic hydrocarbon criteria. I. Water and tissue. Environ Toxicol Chem 19: 1951-1970. |
| [58] |
Kalf DF, Crommentuijn T, van de Plassche EJ (1997) Environmental quality objectives for 10 polycyclic aromatic hydrocarbons (PAHs). Ecotoxicol Environ Saf 36: 89-97. doi: 10.1006/eesa.1996.1495
|
| [59] | Canadian Council of Ministers of the Environment (CCME) (2010) Canadian Soil Quality Guidelines, Carcinogenic and Other Polycyclic Aromatic Hydrocarbons (PAHs)-Environmental and Human Health Effects. ISBN 978-1-896997-94-0 PDF. |
| [60] |
Yang B, Xue N, Zhou L, et al. (2012) Risk assessment and sources of polycyclic aromatic hydrocarbons in agricultural soils of Huanghuai plain, China. Ecotoxicol Environ Saf 84: 304-310. doi: 10.1016/j.ecoenv.2012.07.027
|
| [61] | Omayma EA, Sawsan AM, El Nady MM (2016) Application of polycyclic aromatic hydrocarbons in identification of organic pollution in seawater around Alexandria coastal area, Egypt. J Environ Life Sci 1: 39-55. |
| [62] | Daisey JM, Leyko MA, Kneip TJ (1979) Source identification and allocation of polynuclear aromatic hydrocarbon compounds in the New York City aerosol: methods and applications. In: Jones, P.W., Leber, P. (Eds.), Polynuclear Aromatic Hydrocarbons. Ann Arbor Science, Ann Arbor, pp. 201-215. |
| [63] |
Harrison RM, Smith DJT, Luhana L (1996) Source apportionment of atmospheric polycyclic aromatic hydrocarbons collected from an urban location in Birmingham, UK. Environ Sci Technol 30: 825-832. doi: 10.1021/es950252d
|
| [64] | Rogge WF, Hildemann LM, Mazurek MA, et al. (1993) Source of fine organic aerosol 2. Noncatalyst and catalyst-equipped automobiles and heavy-duty diesel trucks. Environ Sci Technol 27: 636-651. |
| [65] |
Gschwend PM, Hites RA (1981) Fluxes of polycyclic aromatic hydrocarbons to marine and lacustrine sediments in the northeastern United States. Geochimica et Cosmochimica Acta 45: 2359-2367. doi: 10.1016/0016-7037(81)90089-2
|
| [66] |
Mitra S, Bianchi TS, Mckee BA, et al. (2002) Black carbon from the Mississippi River: quantities, sources and potential implications for the global carbon cycle. Environ Sci Technol 36: 2296-2302. doi: 10.1021/es015834b
|
| [67] |
Masclet P, Bresson MA, Mouvier G (1987) Polycyclic aromatic hydrocarbons emitted by power station, and influence of combustion conditions. Fuel 66: 556-562. doi: 10.1016/0016-2361(87)90163-3
|
Figures(2) / Tables(5)

P. U. Singare, J.P. Shirodkar. Persistent and carcinogenic polycyclic aromatic hydrocarbons in the north-western coastal marine environment of India[J]. AIMS Environmental Science, 2021, 8(2): 169-189. doi: 10.3934/environsci.2021012







 DownLoad:
DownLoad: