Citation: Carla Viveiros, Rui Melicio, Victor Mendes, Jose Igreja. Adaptive and predictive controllers applied to onshore wind energy conversion system[J]. AIMS Energy, 2018, 6(4): 615-631. doi: 10.3934/energy.2018.4.615

| [1] | World Wind Energy Association (2018) Wind Power Capacity reaches 539 GW, 52,6 GW added in 2017. Available from: http://www.wwindea.org/2017-statistics/ |
| [2] |
Vidal Y, Acho L, Luo N, et al. (2012) Power control design for variable-speed wind turbines. Energies 5: 3033–3050. doi: 10.3390/en5083033

|
| [3] | Garcia-Sanz M (2012) Wind Energy Systems: Control Engineering Design. CRC Press. |
| [4] | Melicio R, Mendes VMF (2005) Doubly fed induction generator systems for variable speed wind turbine. In Proc. 9th Spanish-Portuguese Congress on Electrical Engineering-9CHLIE, Marbella, Spain, 161–164. |
| [5] | Siraj K, Siraj H, Nasir M (2014) Modeling and control of a doubly fed induction generator for grid integrated wind turbine. In IEEE International Power Electronics and Motion Control Conference and Exposition-PEMC 2014, Antalya, Turkey, 901–906. |
| [6] | Zhang J, Cheng M, Chen,Z, et al. (2008) Pitch angle control for variable speed wind turbines. In 3rd International Conference on Electric Utility Deregulation and Restructuring and Power Technologies-DRPT 2008, Nanjuing, 2691–2696. |
| [7] | Merabet A, Thongam J, Gu J (2011) Torque and pitch angle control for variable speed wind turbines in all operating regimes. In 10th International Conference on Environment and Electrical Engineering-EEEIC 2011, Rome, Italy, 1–5. |
| [8] | Lupu L, Boukhezzar B, Siguerdidjane H (2006) Pitch and torque control strategy for variable speed wind turbines. In European Wind Energy Conference & Exhibition-EWEC 2006, Athens, Greece, 1–7. |
| [9] | Bianchi FD, Battista HD, Mantz RJ (2010) Robust multivariable gain-scheduled control of wind turbines for variable power production. Int J Syst Control 1: 103–112. |
| [10] |
Aissaoui AG, Tahour A, Essounbouli N, et al. (2013) A fuzzy-pi control to extract an optimal power from wind turbine. Energ Convers Manage 65: 688–696. doi: 10.1016/j.enconman.2011.11.034
|
| [11] | Mateescu R, Pintea A, Stefanoiu D (2012) Discrete-time LQG control with disturbance rejection for variable speed wind turbines. In 1st International Conference on Systems and Computer Science-ICSCS 2012, Lille, France, 1–6. |
| [12] |
Simani S, Castaldi P (2013) Data-driven and adaptive control applications to a wind turbine benchmark model. Control Eng Pract 21: 1678–1693. doi: 10.1016/j.conengprac.2013.08.009
|
| [13] |
Beltran B, Ahmed-Ali T, Benbouzid MEH (2008) Sliding mode power control of variable-speed wind energy conversion systems. IEEE T Energy Conver 23: 551–558. doi: 10.1109/TEC.2007.914163
|
| [14] |
Tchakoua P, Wamkeue R, Ouhrouche M, et al. (2014) Wind turbine condition monitoring: State-of-the-art review. Energies 7: 2595–2630. doi: 10.3390/en7042595
|
| [15] | Yang W, Jiang J (2011) Wind turbine condition monitoring and reliability analysis by SCADA information. In IEEE International Conference on Mechanic Automation and Control Engineering-MACE 2011, Hohhot, China, 1872–1875. |
| [16] |
Qi W, Liu J, Chen X, et al. (2011) Supervisory predictive control of standalone wind/solar energy generation systems. IEEE T Contr Syst T 19: 199–207. doi: 10.1109/TCST.2010.2041930
|
| [17] | Sarrias R, Fernández LM, García CA, et al. (2011) Supervisory control system for DFIG wind turbine with energy storage system based on battery. In: International Conference on Power Engineering, Energy and Electrical Drives-POWERENG, Malaga, Spain, 1–6. |
| [18] | Johnson KE, Pao LY, Balas MJ, et al. (2006) Control of variable-speed wind turbines: Standard and adaptive techniques for maximizing energy capture. IEEE Control Syst 26: 70–81. |
| [19] | Odgaard PF, Stroustrup J, Kinnaert M (2013) Fault tolerant control of wind turbines: A benchmark model. IEEE T Contr Syst T 12: 1168–1182. |
| [20] | Melicio R, Mendes VMF, Catalão JPS (2010) Wind turbines equipped with fractional-order controllers: Stress on the mechanical drive train due to a converter control malfunction. Wind Energy 14: 13–25. |
| [21] | Melicio R, Mendes VMF, Catalão JPS (2008) Two-level and multilevel converters for wind energy systems: A comparative study. In Proc. 13th International Power Electronics and Motion Control Conference-EPE-PEMC 2008, Poznań, Poland, 1682–1687. |
| [22] | Melicio R, Mendes VMF, Catalão JPS (2010) Modeling, control and simulation of full-power converter wind turbines equipped with permanent magnet synchronous generator. Int Rev Electr Eng-I 5: 397–408. |
| [23] |
Melicio R, Mendes VMF, Catalão JPS (2009) Modeling and simulation of wind energy systems with matrix and multilevel power converters. IEEE Lat Am T 7: 78–84. doi: 10.1109/TLA.2009.5173468
|
| [24] | William L (2010) The Control Handbook, 2nd ed., Florida: CRC Press. |
| [25] | Maciejowski JM, Goulart PJ, Kerrigan EC (2007) Constrained Control Using Model Predictive Control, In: Tarbouriech S, Garcia G, Glattfelder AH (eds), LNICS, Springer, Heidelberg, 346: 273–291. |
| [26] | Cassandras CG, Lafortune S (2000) Introduction to discrete event systems. Springer Science Business Media, New York, USA. |
| [27] |
Viveiros C, Melicio R, Igreja JM, et al. (2015) Supervisory control of a variable speed wind turbine with doubly fed induction generator. Energ Rep 1: 89–95. doi: 10.1016/j.egyr.2015.03.001
|
| [28] |
Viveiros C, Melicio R, Igreja JM, et al. (2015) Performance assessment of a wind energy conversion system using a hierarchical controller structure. Energ Convers Manage 93: 40–48. doi: 10.1016/j.enconman.2015.01.002
|
| [29] | Viveiros C, Melicio R, Igreja JM, et al. (2013) Application of a discrete adaptive LQG and fuzzy control design to a wind turbine benchmark model. In Proc. 2nd International Conference on Renewable Energy Research and Applications-ICRERA 2013, Madrid, Spain, 488–493. |
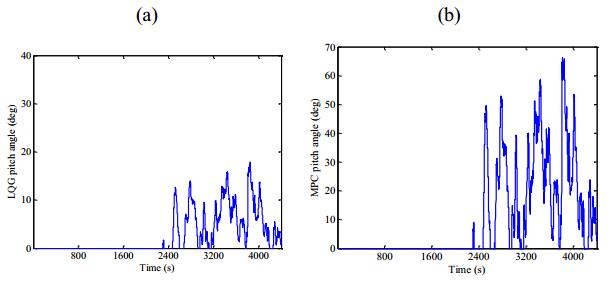
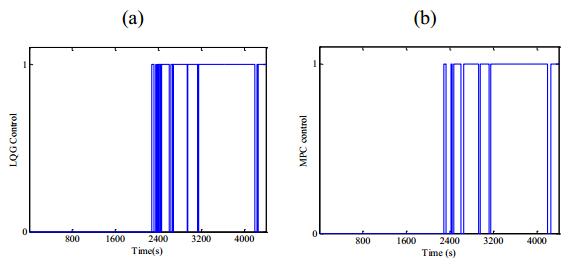
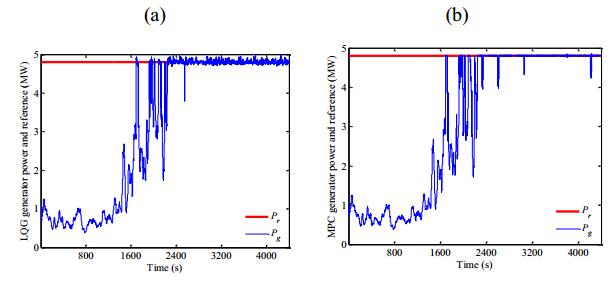
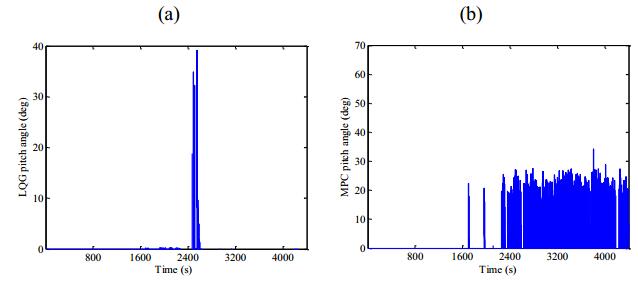
Figures(13) / Tables(1)

Carla Viveiros, Rui Melicio, Victor Mendes, Jose Igreja. Adaptive and predictive controllers applied to onshore wind energy conversion system[J]. AIMS Energy, 2018, 6(4): 615-631. doi: 10.3934/energy.2018.4.615







 DownLoad:
DownLoad: