Citation: Alessandro Didonna, Federico Benetti. Post-translational modifications in neurodegeneration[J]. AIMS Biophysics, 2016, 3(1): 27-49. doi: 10.3934/biophy.2016.1.27

| [1] |
Consortium IHGS (2004) Finishing the euchromatic sequence of the human genome. Nature 431: 931–945. doi: 10.1038/nature03001

|
| [2] |
Yura K, Shionyu M, Hagino K, et al. (2006) Alternative splicing in human transcriptome: functional and structural influence on proteins. Gene 380: 63–71. doi: 10.1016/j.gene.2006.05.015
|
| [3] |
Jensen ON (2004) Modification-specific proteomics: characterization of post-translational modifications by mass spectrometry. Curr Opin Chem Biol 8: 33–41. doi: 10.1016/j.cbpa.2003.12.009
|
| [4] | Khoury GA, Baliban RC, Floudas CA (2011) Proteome-wide post-translational modification statistics: frequency analysis and curation of the swiss-prot database. Sci Rep 1: 90. |
| [5] | Nestler EJ, Greengard P (1999) Protein phosphorylation is of fundamental importance in biological Regulation, In: Siegel G, Agranoff B, Albers R, et al., editors. Basic Neurochemistry: Molecular, Cellular and Medical Aspects. Philadelphia: Lippincott-Raven. |
| [6] |
Hunter T (1995) Protein kinases and phosphatases: the yin and yang of protein phosphorylation and signaling. Cell 80: 225–236. doi: 10.1016/0092-8674(95)90405-0
|
| [7] |
Williams DR (2006) Tauopathies: classification and clinical update on neurodegenerative diseases associated with microtubule-associated protein tau. Intern Med J 36: 652–660. doi: 10.1111/j.1445-5994.2006.01153.x
|
| [8] |
Goedert M, Spillantini MG, Jakes R, et al. (1989) Multiple isoforms of human microtubule-associated protein tau: sequences and localization in neurofibrillary tangles of Alzheimer's disease. Neuron 3: 519–526. doi: 10.1016/0896-6273(89)90210-9
|
| [9] | Mandelkow EM, Mandelkow E (2012) Biochemistry and cell biology of tau protein in neurofibrillary degeneration. Cold Spring Harb Perspect Med 2: a006247. |
| [10] |
Hanger DP, Seereeram A, Noble W (2009) Mediators of tau phosphorylation in the pathogenesis of Alzheimer's disease. Expert Rev Neurother 9: 1647–1666. doi: 10.1586/ern.09.104
|
| [11] |
Gong CX, Iqbal K (2008) Hyperphosphorylation of microtubule-associated protein tau: a promising therapeutic target for Alzheimer disease. Curr Med Chem 15: 2321–2328. doi: 10.2174/092986708785909111
|
| [12] |
Tanimukai H, Grundke-Iqbal I, Iqbal K (2005) Up-regulation of inhibitors of protein phosphatase-2A in Alzheimer's disease. Am J Pathol 166: 1761–1771. doi: 10.1016/S0002-9440(10)62486-8
|
| [13] |
Wang J, Tung YC, Wang Y, et al. (2001) Hyperphosphorylation and accumulation of neurofilament proteins in Alzheimer disease brain and in okadaic acid-treated SY5Y cells. FEBS Lett 507: 81–87. doi: 10.1016/S0014-5793(01)02944-1
|
| [14] |
Ulloa L, Montejo de Garcini E, Gomez-Ramos P, et al. (1994) Microtubule-associated protein MAP1B showing a fetal phosphorylation pattern is present in sites of neurofibrillary degeneration in brains of Alzheimer's disease patients. Brain Res Mol Brain Res 26: 113–122. doi: 10.1016/0169-328X(94)90081-7
|
| [15] | Lindwall G, Cole RD (1984) Phosphorylation affects the ability of tau protein to promote microtubule assembly. J Biol Chem 259: 5301–5305. |
| [16] |
Jeganathan S, Hascher A, Chinnathambi S, et al. (2008) Proline-directed pseudo-phosphorylation at AT8 and PHF1 epitopes induces a compaction of the paperclip folding of Tau and generates a pathological (MC-1) conformation. J Biol Chem 283: 32066–32076. doi: 10.1074/jbc.M805300200
|
| [17] | Tenreiro S, Eckermann K, Outeiro TF (2014) Protein phosphorylation in neurodegeneration: friend or foe? Front Mol Neurosci 7: 42. |
| [18] |
Sato-Harada R, Okabe S, Umeyama T, et al. (1996) Microtubule-associated proteins regulate microtubule function as the track for intracellular membrane organelle transports. Cell Struct Funct 21: 283–295. doi: 10.1247/csf.21.283
|
| [19] |
Cowan CM, Bossing T, Page A, et al. (2010) Soluble hyper-phosphorylated tau causes microtubule breakdown and functionally compromises normal tau in vivo. Acta Neuropathol 120: 593–604. doi: 10.1007/s00401-010-0716-8
|
| [20] |
Hoover BR, Reed MN, Su J, et al. (2010) Tau mislocalization to dendritic spines mediates synaptic dysfunction independently of neurodegeneration. Neuron 68: 1067–1081. doi: 10.1016/j.neuron.2010.11.030
|
| [21] |
Bancher C, Lassmann H, Budka H, et al. (1989) An antigenic profile of Lewy bodies: immunocytochemical indication for protein phosphorylation and ubiquitination. J Neuropathol Exp Neurol 48: 81–93. doi: 10.1097/00005072-198901000-00007
|
| [22] | Thomas B, Beal MF (2007) Parkinson's disease. Hum Mol Genet 16 Spec No. 2: R183–194. |
| [23] |
Bendor JT, Logan TP, Edwards RH (2013) The function of alpha-synuclein. Neuron 79: 1044–1066. doi: 10.1016/j.neuron.2013.09.004
|
| [24] |
Waxman EA, Giasson BI (2011) Characterization of kinases involved in the phosphorylation of aggregated alpha-synuclein. J Neurosci Res 89: 231–247. doi: 10.1002/jnr.22537
|
| [25] |
Anderson JP, Walker DE, Goldstein JM, et al. (2006) Phosphorylation of Ser-129 is the dominant pathological modification of alpha-synuclein in familial and sporadic Lewy body disease. J Biol Chem 281: 29739–29752. doi: 10.1074/jbc.M600933200
|
| [26] |
Paleologou KE, Oueslati A, Shakked G, et al. (2010) Phosphorylation at S87 is enhanced in synucleinopathies, inhibits alpha-synuclein oligomerization, and influences synuclein-membrane interactions. J Neurosci 30: 3184–3198. doi: 10.1523/JNEUROSCI.5922-09.2010
|
| [27] |
Cavallarin N, Vicario M, Negro A (2010) The role of phosphorylation in synucleinopathies: focus on Parkinson's disease. CNS Neurol Disord Drug Targets 9: 471–481. doi: 10.2174/187152710791556140
|
| [28] |
Gorbatyuk OS, Li S, Sullivan LF, et al. (2008) The phosphorylation state of Ser-129 in human alpha-synuclein determines neurodegeneration in a rat model of Parkinson disease. Proc Natl Acad Sci U S A 105: 763–768. doi: 10.1073/pnas.0711053105
|
| [29] |
Kragh CL, Lund LB, Febbraro F, et al. (2009) Alpha-synuclein aggregation and Ser-129 phosphorylation-dependent cell death in oligodendroglial cells. J Biol Chem 284: 10211–10222. doi: 10.1074/jbc.M809671200
|
| [30] | Azeredo da Silveira S, Schneider BL, Cifuentes-Diaz C, et al. (2009) Phosphorylation does not prompt, nor prevent, the formation of alpha-synuclein toxic species in a rat model of Parkinson's disease. Hum Mol Genet 18: 872–887. |
| [31] |
Schreurs S, Gerard M, Derua R, et al. (2014) In vitro phosphorylation does not influence the aggregation kinetics of WT alpha-synuclein in contrast to its phosphorylation mutants. Int J Mol Sci 15: 1040–1067. doi: 10.3390/ijms15011040
|
| [32] |
Xu Y, Deng Y, Qing H (2015) The phosphorylation of alpha-synuclein: development and implication for the mechanism and therapy of the Parkinson's disease. J Neurochem 135: 4–18. doi: 10.1111/jnc.13234
|
| [33] |
Waxman EA, Giasson BI (2008) Specificity and regulation of casein kinase-mediated phosphorylation of alpha-synuclein. J Neuropathol Exp Neurol 67: 402–416. doi: 10.1097/NEN.0b013e3186fc995
|
| [34] |
Goncalves S, Outeiro TF (2013) Assessing the subcellular dynamics of alpha-synuclein using photoactivation microscopy. Mol Neurobiol 47: 1081–1092. doi: 10.1007/s12035-013-8406-x
|
| [35] |
Kontopoulos E, Parvin JD, Feany MB (2006) Alpha-synuclein acts in the nucleus to inhibit histone acetylation and promote neurotoxicity. Hum Mol Genet 15: 3012–3023. doi: 10.1093/hmg/ddl243
|
| [36] |
Scott D, Roy S (2012) alpha-Synuclein inhibits intersynaptic vesicle mobility and maintains recycling-pool homeostasis. J Neurosci 32: 10129–10135. doi: 10.1523/JNEUROSCI.0535-12.2012
|
| [37] |
McFarland MA, Ellis CE, Markey SP, et al. (2008) Proteomics analysis identifies phosphorylation-dependent alpha-synuclein protein interactions. Mol Cell Proteomics 7: 2123–2137. doi: 10.1074/mcp.M800116-MCP200
|
| [38] |
Didonna A, Opal P (2015) The promise and perils of HDAC inhibitors in neurodegeneration. Ann Clin Transl Neurol 2: 79–101. doi: 10.1002/acn3.147
|
| [39] |
Lilja T, Heldring N, Hermanson O (2013) Like a rolling histone: epigenetic regulation of neural stem cells and brain development by factors controlling histone acetylation and methylation. BBA-Gen Subjects 1830: 2354–2360. doi: 10.1016/j.bbagen.2012.08.011
|
| [40] |
Cheung P, Allis CD, Sassone-Corsi P (2000) Signaling to chromatin through histone modifications. Cell 103: 263–271. doi: 10.1016/S0092-8674(00)00118-5
|
| [41] |
Campuzano V, Montermini L, Molto MD, et al. (1996) Friedreich's ataxia: autosomal recessive disease caused by an intronic GAA triplet repeat expansion. Science 271: 1423–1427. doi: 10.1126/science.271.5254.1423
|
| [42] |
Kumari D, Biacsi RE, Usdin K (2011) Repeat expansion affects both transcription initiation and elongation in friedreich ataxia cells. J Biol Chem 286: 4209–4215. doi: 10.1074/jbc.M110.194035
|
| [43] |
Kim E, Napierala M, Dent SY (2011) Hyperexpansion of GAA repeats affects post-initiation steps of FXN transcription in Friedreich's ataxia. Nucleic Acids Res 39: 8366–8377. doi: 10.1093/nar/gkr542
|
| [44] | Pietrobono R, Tabolacci E, Zalfa F, et al. (2005) Molecular dissection of the events leading to inactivation of the FMR1 gene. Hum Mol Genet 14: 267–277. |
| [45] |
Opal P, Zoghbi HY (2002) The role of chaperones in polyglutamine disease. Trends Mol Med 8: 232–236. doi: 10.1016/S1471-4914(02)02310-9
|
| [46] |
Sugars KL, Rubinsztein DC (2003) Transcriptional abnormalities in Huntington disease. Trends Genet 19: 233–238. doi: 10.1016/S0168-9525(03)00074-X
|
| [47] |
Li F, Macfarlan T, Pittman RN, et al. (2002) Ataxin-3 is a histone-binding protein with two independent transcriptional corepressor activities. J Biol Chem 277: 45004–45012. doi: 10.1074/jbc.M205259200
|
| [48] |
Cvetanovic M, Rooney RJ, Garcia JJ, et al. (2007) The role of LANP and ataxin 1 in E4F-mediated transcriptional repression. EMBO Rep 8: 671–677. doi: 10.1038/sj.embor.7400983
|
| [49] |
Venkatraman A, Hu YS, Didonna A, et al. (2014) The histone deacetylase HDAC3 is essential for Purkinje cell function, potentially complicating the use of HDAC inhibitors in SCA1. Hum Mol Genet 23: 3733–3745. doi: 10.1093/hmg/ddu081
|
| [50] |
Hammond JW, Cai D, Verhey KJ (2008) Tubulin modifications and their cellular functions. Curr Opin Cell Biol 20: 71–76. doi: 10.1016/j.ceb.2007.11.010
|
| [51] |
Hubbert C, Guardiola A, Shao R, et al. (2002) HDAC6 is a microtubule-associated deacetylase. Nature 417: 455–458. doi: 10.1038/417455a
|
| [52] |
North BJ, Marshall BL, Borra MT, et al. (2003) The human Sir2 ortholog, SIRT2, is an NAD+-dependent tubulin deacetylase. Mol Cell 11: 437–444. doi: 10.1016/S1097-2765(03)00038-8
|
| [53] |
Chen S, Owens GC, Makarenkova H, et al. (2010) HDAC6 regulates mitochondrial transport in hippocampal neurons. PLoS One 5: e10848. doi: 10.1371/journal.pone.0010848
|
| [54] |
Hempen B, Brion JP (1996) Reduction of acetylated alpha-tubulin immunoreactivity in neurofibrillary tangle-bearing neurons in Alzheimer's disease. J Neuropathol Exp Neurol 55: 964–972. doi: 10.1097/00005072-199609000-00003
|
| [55] |
Dompierre JP, Godin JD, Charrin BC, et al. (2007) Histone deacetylase 6 inhibition compensates for the transport deficit in Huntington's disease by increasing tubulin acetylation. J Neurosci 27: 3571–3583. doi: 10.1523/JNEUROSCI.0037-07.2007
|
| [56] |
d'Ydewalle C, Krishnan J, Chiheb DM, et al. (2011) HDAC6 inhibitors reverse axonal loss in a mouse model of mutant HSPB1-induced Charcot-Marie-Tooth disease. Nat Med 17: 968–974. doi: 10.1038/nm.2396
|
| [57] |
Min SW, Cho SH, Zhou Y, et al. (2010) Acetylation of tau inhibits its degradation and contributes to tauopathy. Neuron 67: 953–966. doi: 10.1016/j.neuron.2010.08.044
|
| [58] |
Min SW, Chen X, Tracy TE, et al. (2015) Critical role of acetylation in tau-mediated neurodegeneration and cognitive deficits. Nat Med 21: 1154–1162. doi: 10.1038/nm.3951
|
| [59] |
Cohen TJ, Guo JL, Hurtado DE, et al. (2011) The acetylation of tau inhibits its function and promotes pathological tau aggregation. Nat Commun 2: 252. doi: 10.1038/ncomms1255
|
| [60] |
Christiansen MN, Chik J, Lee L, et al. (2014) Cell surface protein glycosylation in cancer. Proteomics 14: 525–546. doi: 10.1002/pmic.201300387
|
| [61] |
Spiro RG (2002) Protein glycosylation: nature, distribution, enzymatic formation, and disease implications of glycopeptide bonds. Glycobiology 12: 43R–56R. doi: 10.1093/glycob/12.4.43R
|
| [62] | Aebi M (2013) N-linked protein glycosylation in the ER. BBA-Mol Cell Res 1833: 2430–2437. |
| [63] |
Prusiner SB (1998) Prions. Proc Natl Acad Sci U S A 95: 13363–13383. doi: 10.1073/pnas.95.23.13363
|
| [64] |
Taraboulos A, Rogers M, Borchelt DR, et al. (1990) Acquisition of protease resistance by prion proteins in scrapie-infected cells does not require asparagine-linked glycosylation. Proc Natl Acad Sci U S A 87: 8262–8266. doi: 10.1073/pnas.87.21.8262
|
| [65] |
Lawson VA, Collins SJ, Masters CL, et al. (2005) Prion protein glycosylation. J Neurochem 93: 793–801. doi: 10.1111/j.1471-4159.2005.03104.x
|
| [66] |
Somerville RA (1999) Host and transmissible spongiform encephalopathy agent strain control glycosylation of PrP. J Gen Virol 80: 1865–1872. doi: 10.1099/0022-1317-80-7-1865
|
| [67] |
Safar J, Wille H, Itri V, et al. (1998) Eight prion strains have PrP(Sc) molecules with different conformations. Nat Med 4: 1157–1165. doi: 10.1038/2654
|
| [68] |
Lawson VA, Priola SA, Meade-White K, et al. (2004) Flexible N-terminal region of prion protein influences conformation of protease-resistant prion protein isoforms associated with cross-species scrapie infection in vivo and in vitro. J Biol Chem 279: 13689–13695. doi: 10.1074/jbc.M303697200
|
| [69] |
Steen PVd, Rudd PM, Dwek RA, et al. (1998) Concepts and principles of O-linked glycosylation. Crit Rev Biochem Mol Biol 33: 151–208. doi: 10.1080/10409239891204198
|
| [70] |
Comer FI, Hart GW (2001) Reciprocity between O-GlcNAc and O-phosphate on the carboxyl terminal domain of RNA polymerase II. Biochemistry 40: 7845–7852. doi: 10.1021/bi0027480
|
| [71] |
Lefebvre T, Guinez C, Dehennaut V, et al. (2005) Does O-GlcNAc play a role in neurodegenerative diseases? Expert Rev Proteomics 2: 265–275. doi: 10.1586/14789450.2.2.265
|
| [72] | Robertson LA, Moya KL, Breen KC (2004) The potential role of tau protein O-glycosylation in Alzheimer's disease. J Alzheimers Dis 6: 489–495. |
| [73] |
Mayor S, Riezman H (2004) Sorting GPI-anchored proteins. Nat Rev Mol Cell Biol 5: 110–120. doi: 10.1038/nrm1309
|
| [74] | Taylor DR, Hooper NM (2010) GPI-anchored proteins in health and disease. In: Vidal CJ, editor. Post-translational Modifications in Health and Disease. New York: Springer, 39–55. |
| [75] |
Campana V, Sarnataro D, Zurzolo C (2005) The highways and byways of prion protein trafficking. Trends Cell Biol 15: 102–111. doi: 10.1016/j.tcb.2004.12.002
|
| [76] |
Agostini F, Dotti CG, Perez-Canamas A, et al. (2013) Prion protein accumulation in lipid rafts of mouse aging brain. PLoS One 8: e74244. doi: 10.1371/journal.pone.0074244
|
| [77] |
Taraboulos A, Scott M, Semenov A, et al. (1995) Cholesterol depletion and modification of COOH-terminal targeting sequence of the prion protein inhibit formation of the scrapie isoform. J Cell Biol 129: 121–132. doi: 10.1083/jcb.129.1.121
|
| [78] |
Baron GS, Wehrly K, Dorward DW, et al. (2002) Conversion of raft associated prion protein to the protease-resistant state requires insertion of PrP-res (PrP(Sc)) into contiguous membranes. EMBO J 21: 1031–1040. doi: 10.1093/emboj/21.5.1031
|
| [79] |
Chesebro B, Trifilo M, Race R, et al. (2005) Anchorless prion protein results in infectious amyloid disease without clinical scrapie. Science 308: 1435–1439. doi: 10.1126/science.1110837
|
| [80] |
McNally KL, Ward AE, Priola SA (2009) Cells expressing anchorless prion protein are resistant to scrapie infection. J Virol 83: 4469–4475. doi: 10.1128/JVI.02412-08
|
| [81] |
Munday AD, López JA (2007) Posttranslational protein palmitoylation promoting platelet purpose. Arterioscler Thromb Vasc Biol 27: 1496–1499. doi: 10.1161/ATVBAHA.106.136226
|
| [82] | Resh MD (1999) Fatty acylation of proteins: new insights into membrane targeting of myristoylated and palmitoylated proteins. BBA-Mol Cell Res 1451: 1–16. |
| [83] |
Salaun C, Greaves J, Chamberlain LH (2010) The intracellular dynamic of protein palmitoylation. J Cell Biol 191: 1229–1238. doi: 10.1083/jcb.201008160
|
| [84] |
Zeidman R, Jackson CS, Magee AI (2009) Protein acyl thioesterases (Review). Mol Membr Biol 26: 32–41. doi: 10.1080/09687680802629329
|
| [85] |
Greaves J, Chamberlain LH (2007) Palmitoylation-dependent protein sorting. J Cell Biol 176: 249–254. doi: 10.1083/jcb.200610151
|
| [86] |
Yanai A, Huang K, Kang R, et al. (2006) Palmitoylation of huntingtin by HIP14 is essential for its trafficking and function. Nat Neurosci 9: 824–831. doi: 10.1038/nn1702
|
| [87] |
Bhattacharyya R, Barren C, Kovacs DM (2013) Palmitoylation of amyloid precursor protein regulates amyloidogenic processing in lipid rafts. J Neurosci 33: 11169–11183. doi: 10.1523/JNEUROSCI.4704-12.2013
|
| [88] |
Meckler X, Roseman J, Das P, et al. (2010) Reduced Alzheimer's disease ss-amyloid deposition in transgenic mice expressing S-palmitoylation-deficient APH1aL and nicastrin. J Neurosci 30: 16160–16169. doi: 10.1523/JNEUROSCI.4436-10.2010
|
| [89] |
Farazi TA, Waksman G, Gordon JI (2001) The biology and enzymology of protein N-myristoylation. J Biol Chem 276: 39501–39504. doi: 10.1074/jbc.R100042200
|
| [90] |
Martin DD, Ahpin CY, Heit RJ, et al. (2012) Tandem reporter assay for myristoylated proteins post-translationally (TRAMPP) identifies novel substrates for post-translational myristoylation: PKCepsilon, a case study. FASEB J 26: 13–28. doi: 10.1096/fj.11-182360
|
| [91] |
Martin DD, Heit RJ, Yap MC, et al. (2014) Identification of a post-translationally myristoylated autophagy-inducing domain released by caspase cleavage of huntingtin. Hum Mol Genet 23: 3166–3179. doi: 10.1093/hmg/ddu027
|
| [92] |
Martin DD, Ladha S, Ehrnhoefer DE, et al. (2015) Autophagy in Huntington disease and huntingtin in autophagy. Trends Neurosci 38: 26–35. doi: 10.1016/j.tins.2014.09.003
|
| [93] |
Ciechanover A (2005) Intracellular protein degradation: from a vague idea thru the lysosome and the ubiquitin–proteasome system and onto human diseases and drug targeting. Cell Death Differ 12: 1178–1190. doi: 10.1038/sj.cdd.4401692
|
| [94] |
Mukhopadhyay D, Riezman H (2007) Proteasome-independent functions of ubiquitin in endocytosis and signaling. Science 315: 201–205. doi: 10.1126/science.1127085
|
| [95] |
Schnell JD, Hicke L (2003) Non-traditional functions of ubiquitin and ubiquitin-binding proteins. J Biol Chem 278: 35857–35860. doi: 10.1074/jbc.R300018200
|
| [96] |
Zeng L-R, Vega-Sánchez ME, Zhu T, et al. (2006) Ubiquitination-mediated protein degradation and modification: an emerging theme in plant-microbe interactions. Cell Res 16: 413–426. doi: 10.1038/sj.cr.7310053
|
| [97] |
Marx J (2002) Ubiquitin Lives Up to Its Name. Science 297: 1792–1794 doi: 10.1126/science.297.5588.1792
|
| [98] | Schipper-Krom S, Juenemann K, Reits EA (2012) The ubiquitin-proteasome system in Huntington’s disease: are proteasomes impaired, initiators of disease, or coming to the rescue? Biochem Res Int 2012: 1–12. |
| [99] |
Mitra S, Finkbeiner S (2008) The ubiquitin-proteasome pathway in Huntington's disease. Sci World J 8: 421–433. doi: 10.1100/tsw.2008.60
|
| [100] |
Kalchman MA, Graham RK, Xia G, et al. (1996) Huntingtin is ubiquitinated and interacts with a specific ubiquitin-conjugating enzyme. J Biol Chem 271: 19385–19394. doi: 10.1074/jbc.271.32.19385
|
| [101] |
Venkatraman P, Wetzel R, Tanaka M, et al. (2004) Eukaryotic proteasomes cannot digest polyglutamine sequences and release them during degradation of polyglutamine-containing proteins. Mol Cell 14: 95–104. doi: 10.1016/S1097-2765(04)00151-0
|
| [102] |
Suhr ST, Senut M-C, Whitelegge JP, et al. (2001) Identities of sequestered proteins in aggregates from cells with induced polyglutamine expression. J Cell Biol 153: 283–294. doi: 10.1083/jcb.153.2.283
|
| [103] |
Seo H, Sonntag K-C, Kim W, et al. (2007) Proteasome activator enhances survival of Huntington’s disease neuronal model cells. PloS one 2: e238. doi: 10.1371/journal.pone.0000238
|
| [104] |
Jia H, Kast RJ, Steffan JS, et al. (2012) Selective histone deacetylase (HDAC) inhibition imparts beneficial effects in Huntington's disease mice: implications for the ubiquitin–proteasomal and autophagy systems. Hum Mol Genet 21: 5280–5293. doi: 10.1093/hmg/dds379
|
| [105] |
Perry G, Friedman R, Shaw G, et al. (1987) Ubiquitin is detected in neurofibrillary tangles and senile plaque neurites of Alzheimer disease brains. Proc Natl Acad Sci U S A 84: 3033–3036. doi: 10.1073/pnas.84.9.3033
|
| [106] |
Salon ML, Pasquini L, Moreno MB, et al. (2003) Relationship between β-amyloid degradation and the 26S proteasome in neural cells. Exp Neurol 180: 131–143. doi: 10.1016/S0014-4886(02)00060-2
|
| [107] |
Almeida CG, Takahashi RH, Gouras GK (2006) β-Amyloid accumulation impairs multivesicular body sorting by inhibiting the ubiquitin-proteasome system. J Neurosci 26: 4277–4288. doi: 10.1523/JNEUROSCI.5078-05.2006
|
| [108] |
Zhao X, Yang J (2010) Amyloid-β peptide is a substrate of the human 20S proteasome. ACS Chem Neurosci 1: 655–660. doi: 10.1021/cn100067e
|
| [109] |
Van Leeuwen FW, de Kleijn DP, van den Hurk HH, et al. (1998) Frameshift mutants of β amyloid precursor protein and ubiquitin-B in Alzheimer's and Down patients. Science 279: 242–247. doi: 10.1126/science.279.5348.242
|
| [110] |
Chadwick L, Gentle L, Strachan J, et al. (2012) Review: Unchained maladie–a reassessment of the role of Ubb+ 1‐capped polyubiquitin chains in Alzheimer's disease. Neuropathol App Neurobiol 38: 118–131. doi: 10.1111/j.1365-2990.2011.01236.x
|
| [111] |
Lam YA, Pickart CM, Alban A, et al. (2000) Inhibition of the ubiquitin-proteasome system in Alzheimer's disease. Proc Natl Acad Sci U S A 97: 9902–9906. doi: 10.1073/pnas.170173897
|
| [112] |
Wilkinson K, Henley J (2010) Mechanisms, regulation and consequences of protein SUMOylation. Biochem J 428: 133–145. doi: 10.1042/BJ20100158
|
| [113] |
Riley BE, Zoghbi HY, Orr HT (2005) SUMOylation of the polyglutamine repeat protein, ataxin-1, is dependent on a functional nuclear localization signal. J Biol Chem 280: 21942–21948. doi: 10.1074/jbc.M501677200
|
| [114] |
Steffan JS, Agrawal N, Pallos J, et al. (2004) SUMO modification of Huntingtin and Huntington's disease pathology. Science 304: 100–104. doi: 10.1126/science.1092194
|
| [115] |
Poukka H, Karvonen U, Jänne OA, et al. (2000) Covalent modification of the androgen receptor by small ubiquitin-like modifier 1 (SUMO-1). Proc Natl Acad Sci U S A 97: 14145–14150. doi: 10.1073/pnas.97.26.14145
|
| [116] |
Chua JP, Reddy SL, Yu Z, et al. (2015) Disrupting SUMOylation enhances transcriptional function and ameliorates polyglutamine androgen receptor–mediated disease. J Clin Invest 125: 831–845. doi: 10.1172/JCI73214
|
| [117] |
Terashima T, Kawai H, Fujitani M, et al. (2002) SUMO-1 co-localized with mutant atrophin-1 with expanded polyglutamines accelerates intranuclear aggregation and cell death. Neuroreport 13: 2359–2364. doi: 10.1097/00001756-200212030-00038
|
| [118] | Geiger T, Clarke S (1987) Deamidation, isomerization, and racemization at asparaginyl and aspartyl residues in peptides. Succinimide-linked reactions that contribute to protein degradation. J Biol Chem 262: 785–794. |
| [119] |
Yang H, Zubarev RA (2010) Mass spectrometric analysis of asparagine deamidation and aspartate isomerization in polypeptides. Electrophoresis 31: 1764–1772. doi: 10.1002/elps.201000027
|
| [120] |
Robinson AB, McKerrow JH, Cary P (1970) Controlled deamidation of peptides and proteins: an experimental hazard and a possible biological timer. Proc Natl Acad Sci U S A 66: 753–757. doi: 10.1073/pnas.66.3.753
|
| [121] | Watanabe A, Takio K, Ihara Y (1999) Deamidation and isoaspartate formation in smeared tau in paired helical filaments. Unusual properties of the microtubule-binding domain of tau. J Biol Chem 274: 7368–7378. |
| [122] |
Dan A, Takahashi M, Masuda-Suzukake M, et al. (2013) Extensive deamidation at asparagine residue 279 accounts for weak immunoreactivity of tau with RD4 antibody in Alzheimer's disease brain. Acta Neuropathol Commun 1: 54. doi: 10.1186/2051-5960-1-54
|
| [123] |
Shimizu T, Watanabe A, Ogawara M, et al. (2000) Isoaspartate formation and neurodegeneration in Alzheimer's disease. Arch Biochem Biophys 381: 225–234. doi: 10.1006/abbi.2000.1955
|
| [124] | Yamamoto A, Takagi H, Kitamura D, et al. (1998) Deficiency in protein L-isoaspartyl methyltransferase results in a fatal progressive epilepsy. J Neurosci 18: 2063–2074. |
| [125] |
O'Brien JT, Thomas A (2015) Vascular dementia. Lancet 386: 1698–1706. doi: 10.1016/S0140-6736(15)00463-8
|
| [126] |
Adav SS, Qian J, Ang YL, et al. (2014) iTRAQ quantitative clinical proteomics revealed role of Na(+)K(+)-ATPase and its correlation with deamidation in vascular dementia. J Proteome Res 13: 4635–4646. doi: 10.1021/pr500754j
|
| [127] |
Moss CX, Matthews SP, Lamont DJ, et al. (2005) Asparagine deamidation perturbs antigen presentation on class II major histocompatibility complex molecules. J Biol Chem 280: 18498–18503. doi: 10.1074/jbc.M501241200
|
| [128] |
Manoury B, Mazzeo D, Fugger L, et al. (2002) Destructive processing by asparagine endopeptidase limits presentation of a dominant T cell epitope in MBP. Nat Immunol 3: 169–174. doi: 10.1038/ni754
|
| [129] | Kim JK, Mastronardi FG, Wood DD, et al. (2003) Multiple sclerosis: an important role for post-translational modifications of myelin basic protein in pathogenesis. Mol Cell Proteomics 2: 453–462. |
| [130] | Sayre LM, Moreira PI, Smith MA, et al. (2005) Metal ions and oxidative protein modification in neurological disease. Ann Ist Super Sanita 41: 143–164. |
| [131] |
Stadtman ER (1992) Protein oxidation and aging. Science 257: 1220–1224. doi: 10.1126/science.1355616
|
| [132] | Cai Z, Yan L-J (2013) Protein oxidative modifications: beneficial roles in disease and health. J Biochem Pharmacol Res 1: 15–26. |
| [133] |
Sayre LM, Perry G, Smith MA (1999) Redox metals and neurodegenerative disease. Curr Opin Chem Biol 3: 220–225. doi: 10.1016/S1367-5931(99)80035-0
|
| [134] | Sayre LM, Perry G, Atwood CS, et al. (2000) The role of metals in neurodegenerative diseases. Cell Mol Biol (Noisy-le-grand) 46: 731–741. |
| [135] |
Uttara B, Singh AV, Zamboni P, et al. (2009) Oxidative stress and neurodegenerative diseases: a review of upstream and downstream antioxidant therapeutic options. Curr Neuropharmacol 7: 65–74. doi: 10.2174/157015909787602823
|
| [136] |
Gasperini L, Meneghetti E, Pastore B, et al. (2015) Prion protein and copper cooperatively protect neurons by modulating NMDA receptor through S-nitrosylation. Antioxid Redox Signal 22: 772–784. doi: 10.1089/ars.2014.6032
|
| [137] | Stadtman ER (2001) Protein oxidation in aging and age‐related diseases. Ann N Y Acad Sci 928: 22–38. |
| [138] | Bizzozero OA (2009) Protein carbonylation in neurodegenerative and demyelinating CNS diseases. In: Lajtha A, Banik N, Ray S, editors. Handbook of Neurochemistry and Molecular Neurobiology. New York: Springer, 543–462. |
| [139] |
Wong CM, Cheema AK, Zhang L, et al. (2008) Protein carbonylation as a novel mechanism in redox signaling. Circ Res 102: 310–318. doi: 10.1161/CIRCRESAHA.107.159814
|
| [140] |
Wong C-M, Bansal G, Marcocci L, et al. (2012) Proposed role of primary protein carbonylation in cell signaling. Redox Rep 17: 90–94. doi: 10.1179/1351000212Y.0000000007
|
| [141] |
Serviddio G, Di Venosa N, Federici A, et al. (2005) Brief hypoxia before normoxic reperfusion (postconditioning) protects the heart against ischemia-reperfusion injury by preventing mitochondria peroxyde production and glutathione depletion. FASEB J 19: 354–361. doi: 10.1096/fj.04-2338com
|
| [142] |
Lee JR, Kim JK, Lee SJ, et al. (2009) Role of protein tyrosine nitration in neurodegenerative diseases and atherosclerosis. Arch Pharm Res 32: 1109–1118. doi: 10.1007/s12272-009-1802-0
|
| [143] |
Reynolds MR, Reyes JF, Fu Y, et al. (2006) Tau nitration occurs at tyrosine 29 in the fibrillar lesions of Alzheimer's disease and other tauopathies. J Neurosci 26: 10636–10645. doi: 10.1523/JNEUROSCI.2143-06.2006
|
| [144] |
Reynolds MR, Berry RW, Binder LI (2005) Site-specific nitration differentially influences tau assembly in vitro. Biochemistry 44: 13997–14009. doi: 10.1021/bi051028w
|
| [145] |
Takahashi T, Yamashita H, Nakamura T, et al. (2002) Tyrosine 125 of alpha-synuclein plays a critical role for dimerization following nitrative stress. Brain Res 938: 73–80. doi: 10.1016/S0006-8993(02)02498-8
|
| [146] |
Hodara R, Norris EH, Giasson BI, et al. (2004) Functional consequences of alpha-synuclein tyrosine nitration: diminished binding to lipid vesicles and increased fibril formation. J Biol Chem 279: 47746–47753. doi: 10.1074/jbc.M408906200
|
| [147] |
Giasson BI, Duda JE, Murray IV, et al. (2000) Oxidative damage linked to neurodegeneration by selective alpha-synuclein nitration in synucleinopathy lesions. Science 290: 985–989. doi: 10.1126/science.290.5493.985
|
| [148] | Benhar M, Forrester MT, Stamler JS (2009) Protein denitrosylation: enzymatic mechanisms and cellular functions. Nat Rev Mol Cell Biol 10: 721–732. |
| [149] |
Nakamura T, Tu S, Akhtar MW, et al. (2013) Aberrant protein s-nitrosylation in neurodegenerative diseases. Neuron 78: 596–614. doi: 10.1016/j.neuron.2013.05.005
|
| [150] |
Zhao Q-F, Yu J-T, Tan L (2015) S-Nitrosylation in Alzheimer's disease. Mol Neurobiol 51: 268–280. doi: 10.1007/s12035-014-8672-2
|
| [151] | Chung K, Dawson TM, Dawson VL (2005) Nitric oxide, S-nitrosylation and neurodegeneration. Cell Mol Biol (Noisy-le-Grand, France) 51: 247–254. |
| [152] |
Maiti NR, Surewicz WK (2001) The role of disulfide bridge in the folding and stability of the recombinant human prion protein. J Biol Chem 276: 2427–2431. doi: 10.1074/jbc.M007862200
|
| [153] | Ning L, Guo J, Jin N, et al. (2014) The role of Cys179–Cys214 disulfide bond in the stability and folding of prion protein: insights from molecular dynamics simulations. J Mol Model 20: 1–8. |
| [154] |
Benetti F, Biarnés X, Attanasio F, et al. (2014) Structural determinants in prion protein folding and stability. J Mol Biol 426: 3796–3810. doi: 10.1016/j.jmb.2014.09.017
|
| [155] |
Benetti F, Legname G (2015) New insights into structural determinants of prion protein folding and stability. Prion 9: 119–124. doi: 10.1080/19336896.2015.1022023
|
| [156] |
Shimura H, Hattori N, Kubo S-i, et al. (2000) Familial Parkinson disease gene product, parkin, is a ubiquitin-protein ligase. Nat Genet 25: 302–305. doi: 10.1038/77060
|
| [157] |
Andreu CI, Woehlbier U, Torres M, et al. (2012) Protein disulfide isomerases in neurodegeneration: from disease mechanisms to biomedical applications. FEBS Lett 586: 2826–2834. doi: 10.1016/j.febslet.2012.07.023
|
| [158] |
Smirnova E, Griparic L, Shurland D-L, et al. (2001) Dynamin-related protein Drp1 is required for mitochondrial division in mammalian cells. Mol Bio Cell 12: 2245–2256. doi: 10.1091/mbc.12.8.2245
|
| [159] |
Ohshima T, Ward JM, Huh C-G, et al. (1996) Targeted disruption of the cyclin-dependent kinase 5 gene results in abnormal corticogenesis, neuronal pathology and perinatal death. Proc Natl Acad Sci U S A 93: 11173–11178. doi: 10.1073/pnas.93.20.11173
|
| [160] |
Rhee SG, Kang SW, Jeong W, et al. (2005) Intracellular messenger function of hydrogen peroxide and its regulation by peroxiredoxins. Curr Opin Cell Biol 17: 183–189. doi: 10.1016/j.ceb.2005.02.004
|
| [161] |
Kim Y-S, Joh TH (2012) Matrix metalloproteinases, new insights into the understanding of neurodegenerative disorders. Biomol Ther 20: 133–143. doi: 10.4062/biomolther.2012.20.2.133
|
| [162] |
Chandrasekharan N, Simmons DL (2004) The cyclooxygenases. Genome Biol 5: 241. doi: 10.1186/gb-2004-5-9-241
|
| [163] |
Tristan C, Shahani N, Sedlak TW, et al. (2011) The diverse functions of GAPDH: views from different subcellular compartments. Cell Signal 23: 317–323. doi: 10.1016/j.cellsig.2010.08.003
|
| [164] | Ariga H, Takahashi-Niki K, Kato I, et al. (2013) Neuroprotective function of DJ-1 in Parkinson’s disease. Oxid Med Cell Longev 2013: 1–9. |
| [165] |
Stroissnigg H, Trančíková A, Descovich L, et al. (2007) S-nitrosylation of microtubule-associated protein 1B mediates nitric-oxide-induced axon retraction. Nat Cell Biol 9: 1035–1045. doi: 10.1038/ncb1625
|
| [166] |
Tsang AH, Lee Y-I, Ko HS, et al. (2009) S-nitrosylation of XIAP compromises neuronal survival in Parkinson's disease. Proc Natl Acad Sci U S A 106: 4900–4905. doi: 10.1073/pnas.0810595106
|
| [167] |
Numajiri N, Takasawa K, Nishiya T, et al. (2011) On–off system for PI3-kinase–Akt signaling through S-nitrosylation of phosphatase with sequence homology to tensin (PTEN). Proc Natl Acad Sci U S A 108: 10349–10354. doi: 10.1073/pnas.1103503108
|
| [168] | Zito K, Scheuss V (2009) NMDA receptor function and physiological modulation. In: Squire, LR, editor. The New Encyclopedia of Neuroscience. Oxford: Academic Press, 1157–1164. |
| [169] | Prabakaran S, Lippens G, Steen H, et al. (2012) Post‐translational modification: nature's escape from genetic imprisonment and the basis for dynamic information encoding. Wiley Interdiscip Rev Syst Biol Med 4: 565–583. |
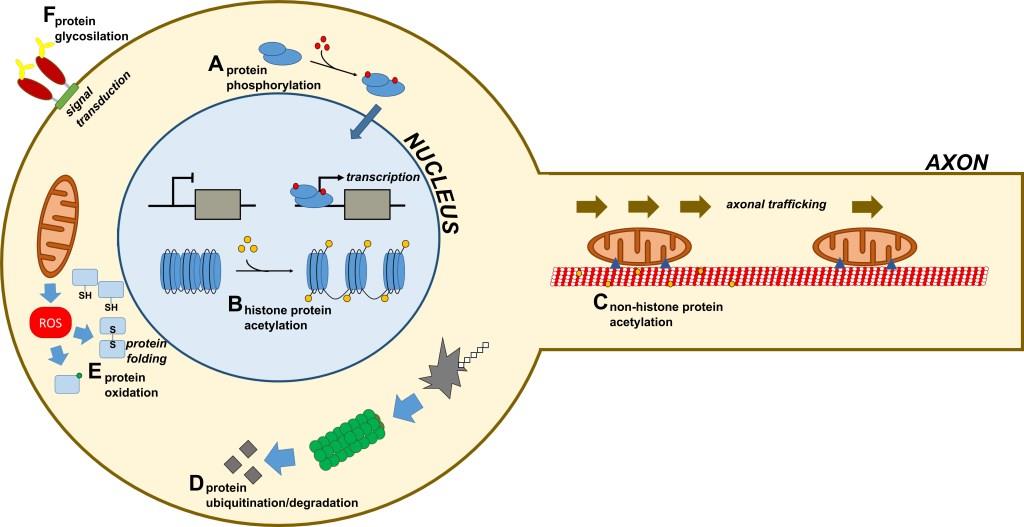
Figures(1) / Tables(1)

Alessandro Didonna, Federico Benetti. Post-translational modifications in neurodegeneration[J]. AIMS Biophysics, 2016, 3(1): 27-49. doi: 10.3934/biophy.2016.1.27







 DownLoad:
DownLoad: