Citation: Andrea Bianchi, Chiara Lanzuolo. Into the chromatin world: Role of nuclear architecture in epigenome regulation[J]. AIMS Biophysics, 2015, 2(4): 585-612. doi: 10.3934/biophy.2015.4.585

| [1] | Luger K, Mader AW, Richmond RK, et al. (1997) Crystal structure of the nucleosome core particle at 2.8 A resolution. Nature 389: 251-260. |
| [2] |
Travers A (1999) The location of the linker histone on the nucleosome. Trends Biochem Sci 24: 4-7. doi: 10.1016/S0968-0004(98)01339-5

|
| [3] | Goytisolo FA, Gerchman SE, Yu X, et al. (1996) Identification of two DNA-binding sites on the globular domain of histone H5. Embo J 15: 3421-3429. |
| [4] |
Thoma F, Koller T, Klug A (1979) Involvement of histone H1 in the organization of the nucleosome and of the salt-dependent superstructures of chromatin. J Cell Biol 83: 403-427. doi: 10.1083/jcb.83.2.403
|
| [5] |
Woodcock CL, Skoultchi AI, Fan Y (2006) Role of linker histone in chromatin structure and function: H1 stoichiometry and nucleosome repeat length. Chromosome Res 14: 17-25. doi: 10.1007/s10577-005-1024-3
|
| [6] |
Fan Y, Nikitina T, Morin-Kensicki EM, et al. (2003) H1 linker histones are essential for mouse development and affect nucleosome spacing in vivo. Mol Cell Biol 23: 4559-4572. doi: 10.1128/MCB.23.13.4559-4572.2003
|
| [7] | Zacharias H (1995) Emil Heitz (1892-1965): chloroplasts, heterochromatin, and polytene chromosomes. Genetics 141: 7-14. |
| [8] |
Huisinga KL, Brower-Toland B, Elgin SC (2006) The contradictory definitions of heterochromatin: transcription and silencing. Chromosoma 115: 110-122. doi: 10.1007/s00412-006-0052-x
|
| [9] |
Gilbert N, Boyle S, Fiegler H, et al. (2004) Chromatin architecture of the human genome: gene-rich domains are enriched in open chromatin fibers. Cell 118: 555-566. doi: 10.1016/j.cell.2004.08.011
|
| [10] |
Gorisch SM, Wachsmuth M, Toth KF, et al. (2005) Histone acetylation increases chromatin accessibility. J Cell Sci 118: 5825-5834. doi: 10.1242/jcs.02689
|
| [11] |
Vakoc CR, Mandat SA, Olenchock BA, et al. (2005) Histone H3 lysine 9 methylation and HP1gamma are associated with transcription elongation through mammalian chromatin. Mol Cell 19: 381-391. doi: 10.1016/j.molcel.2005.06.011
|
| [12] |
Gilbert N, Allan J (2001) Distinctive higher-order chromatin structure at mammalian centromeres. Proc Natl Acad Sci U S A 98: 11949-11954. doi: 10.1073/pnas.211322798
|
| [13] |
Blasco MA (2007) The epigenetic regulation of mammalian telomeres. Nat Rev Genet 8: 299-309. doi: 10.1038/nrg2047
|
| [14] | Slotkin RK, Martienssen R (2007) Transposable elements and the epigenetic regulation of the genome. Nat Rev Genet 8: 272-285. |
| [15] |
Kouzarides T (2007) Chromatin modifications and their function. Cell 128: 693-705. doi: 10.1016/j.cell.2007.02.005
|
| [16] |
Bernstein BE, Kamal M, Lindblad-Toh K, et al. (2005) Genomic maps and comparative analysis of histone modifications in human and mouse. Cell 120: 169-181. doi: 10.1016/j.cell.2005.01.001
|
| [17] | Allshire RC, Ekwall K (2015) Epigenetic Regulation of Chromatin States in Schizosaccharomyces pombe. Cold Spring Harb Perspect Biol 7. |
| [18] |
Nakamura T, Mori T, Tada S, et al. (2002) ALL-1 is a histone methyltransferase that assembles a supercomplex of proteins involved in transcriptional regulation. Mol Cell 10: 1119-1128. doi: 10.1016/S1097-2765(02)00740-2
|
| [19] |
Milne TA, Briggs SD, Brock HW, et al. (2002) MLL targets SET domain methyltransferase activity to Hox gene promoters. Mol Cell 10: 1107-1117. doi: 10.1016/S1097-2765(02)00741-4
|
| [20] |
Trojer P, Reinberg D (2007) Facultative heterochromatin: is there a distinctive molecular signature? Mol Cell 28: 1-13. doi: 10.1016/j.molcel.2007.09.011
|
| [21] |
Rea S, Eisenhaber F, O'Carroll D, et al. (2000) Regulation of chromatin structure by site-specific histone H3 methyltransferases. Nature 406: 593-599. doi: 10.1038/35020506
|
| [22] |
Lachner M, O'Carroll D, Rea S, et al. (2001) Methylation of histone H3 lysine 9 creates a binding site for HP1 proteins. Nature 410: 116-120. doi: 10.1038/35065132
|
| [23] |
Cowell IG, Aucott R, Mahadevaiah SK, et al. (2002) Heterochromatin, HP1 and methylation at lysine 9 of histone H3 in animals. Chromosoma 111: 22-36. doi: 10.1007/s00412-002-0182-8
|
| [24] |
Fanti L, Berloco M, Piacentini L, et al. (2003) Chromosomal distribution of heterochromatin protein 1 (HP1) in Drosophila: a cytological map of euchromatic HP1 binding sites. Genetica 117: 135-147. doi: 10.1023/A:1022971407290
|
| [25] |
Nielsen SJ, Schneider R, Bauer UM, et al. (2001) Rb targets histone H3 methylation and HP1 to promoters. Nature 412: 561-565. doi: 10.1038/35087620
|
| [26] | Lanzuolo C, Orlando V (2012) Memories from the Polycomb Group Proteins. Annu Rev Genet. |
| [27] |
Simon JA, Kingston RE (2009) Mechanisms of polycomb gene silencing: knowns and unknowns. Nat Rev Mol Cell Biol 10: 697-708. doi: 10.1038/nrn2731
|
| [28] |
Boyer LA, Plath K, Zeitlinger J, et al. (2006) Polycomb complexes repress developmental regulators in murine embryonic stem cells. Nature 441: 349-353. doi: 10.1038/nature04733
|
| [29] |
Sauvageau M, Sauvageau G (2010) Polycomb group proteins: multi-faceted regulators of somatic stem cells and cancer. Cell Stem Cell 7: 299-313. doi: 10.1016/j.stem.2010.08.002
|
| [30] |
Zhou W, Zhu P, Wang J, et al. (2008) Histone H2A monoubiquitination represses transcription by inhibiting RNA polymerase II transcriptional elongation. Mol Cell 29: 69-80. doi: 10.1016/j.molcel.2007.11.002
|
| [31] | van Kruijsbergen I, Hontelez S, Veenstra GJ (2015) Recruiting polycomb to chromatin. Int J Biochem Cell Biol. |
| [32] |
Blackledge NP, Farcas AM, Kondo T, et al. (2014) Variant PRC1 complex-dependent H2A ubiquitylation drives PRC2 recruitment and polycomb domain formation. Cell 157: 1445-1459. doi: 10.1016/j.cell.2014.05.004
|
| [33] |
Ku M, Koche RP, Rheinbay E, et al. (2008) Genomewide analysis of PRC1 and PRC2 occupancy identifies two classes of bivalent domains. PLoS Genet 4: e1000242. doi: 10.1371/journal.pgen.1000242
|
| [34] |
Gao Z, Zhang J, Bonasio R, et al. (2012) PCGF Homologs, CBX Proteins, and RYBP Define Functionally Distinct PRC1 Family Complexes. Mol Cell 45: 344-356. doi: 10.1016/j.molcel.2012.01.002
|
| [35] |
Cooper S, Dienstbier M, Hassan R, et al. (2014) Targeting polycomb to pericentric heterochromatin in embryonic stem cells reveals a role for H2AK119u1 in PRC2 recruitment. Cell Rep 7: 1456-1470. doi: 10.1016/j.celrep.2014.04.012
|
| [36] |
Kalb R, Latwiel S, Baymaz HI, et al. (2014) Histone H2A monoubiquitination promotes histone H3 methylation in Polycomb repression. Nat Struct Mol Biol 21: 569-571. doi: 10.1038/nsmb.2833
|
| [37] |
Nazer E, Lei EP (2014) Modulation of chromatin modifying complexes by noncoding RNAs in trans. Curr Opin Genet Dev 25: 68-73. doi: 10.1016/j.gde.2013.11.019
|
| [38] |
Jacinto FV, Benner C, Hetzer MW (2015) The nucleoporin Nup153 regulates embryonic stem cell pluripotency through gene silencing. Genes Dev 29: 1224-1238. doi: 10.1101/gad.260919.115
|
| [39] |
Mozzetta C, Pontis J, Fritsch L, et al. (2014) The histone H3 lysine 9 methyltransferases G9a and GLP regulate polycomb repressive complex 2-mediated gene silencing. Mol Cell 53: 277-289. doi: 10.1016/j.molcel.2013.12.005
|
| [40] |
Sarma K, Cifuentes-Rojas C, Ergun A, et al. (2014) ATRX directs binding of PRC2 to Xist RNA and Polycomb targets. Cell 159: 869-883. doi: 10.1016/j.cell.2014.10.019
|
| [41] |
Wu S, Shi Y, Mulligan P, et al. (2007) A YY1-INO80 complex regulates genomic stability through homologous recombination-based repair. Nat Struct Mol Biol 14: 1165-1172. doi: 10.1038/nsmb1332
|
| [42] |
Margueron R, Justin N, Ohno K, et al. (2009) Role of the polycomb protein EED in the propagation of repressive histone marks. Nature 461: 762-767. doi: 10.1038/nature08398
|
| [43] |
Xu C, Bian C, Yang W, et al. (2010) Binding of different histone marks differentially regulates the activity and specificity of polycomb repressive complex 2 (PRC2). Proc Natl Acad Sci U S A 107: 19266-19271. doi: 10.1073/pnas.1008937107
|
| [44] | Posfai E, Kunzmann R, Brochard V, et al. (2012) Polycomb function during oogenesis is required for mouse embryonic development. Genes Dev. |
| [45] |
Surface LE, Thornton SR, Boyer LA (2010) Polycomb group proteins set the stage for early lineage commitment. Cell Stem Cell 7: 288-298. doi: 10.1016/j.stem.2010.08.004
|
| [46] |
Bracken AP, Dietrich N, Pasini D, et al. (2006) Genome-wide mapping of Polycomb target genes unravels their roles in cell fate transitions. Genes Dev 20: 1123-1136. doi: 10.1101/gad.381706
|
| [47] |
Asp P, Blum R, Vethantham V, et al. (2011) Genome-wide remodeling of the epigenetic landscape during myogenic differentiation. Proc Natl Acad Sci U S A 108: E149-158. doi: 10.1073/pnas.1102223108
|
| [48] |
Caretti G, Di Padova M, Micales B, et al. (2004) The Polycomb Ezh2 methyltransferase regulates muscle gene expression and skeletal muscle differentiation. Genes Dev 18: 2627-2638. doi: 10.1101/gad.1241904
|
| [49] |
Juan AH, Derfoul A, Feng X, et al. (2011) Polycomb EZH2 controls self-renewal and safeguards the transcriptional identity of skeletal muscle stem cells. Genes Dev 25: 789-794. doi: 10.1101/gad.2027911
|
| [50] |
Juan AH, Kumar RM, Marx JG, et al. (2009) Mir-214-dependent regulation of the polycomb protein Ezh2 in skeletal muscle and embryonic stem cells. Mol Cell 36: 61-74. doi: 10.1016/j.molcel.2009.08.008
|
| [51] |
Palacios D, Mozzetta C, Consalvi S, et al. (2010) TNF/p38alpha/polycomb signaling to Pax7 locus in satellite cells links inflammation to the epigenetic control of muscle regeneration. Cell Stem Cell 7: 455-469. doi: 10.1016/j.stem.2010.08.013
|
| [52] | Pombo A, Dillon N (2015) Three-dimensional genome architecture: players and mechanisms. Nat Rev Mol Cell Biol 16: 245-257. |
| [53] |
Bantignies F, Cavalli G (2011) Polycomb group proteins: repression in 3D. Trends Genet 27: 454-464. doi: 10.1016/j.tig.2011.06.008
|
| [54] |
Dekker J, Rippe K, Dekker M, et al. (2002) Capturing chromosome conformation. Science 295: 1306-1311. doi: 10.1126/science.1067799
|
| [55] |
de Wit E, de Laat W (2012) A decade of 3C technologies: insights into nuclear organization. Genes Dev 26: 11-24. doi: 10.1101/gad.179804.111
|
| [56] |
Simonis M, Klous P, Splinter E, et al. (2006) Nuclear organization of active and inactive chromatin domains uncovered by chromosome conformation capture-on-chip (4C). Nat Genet 38: 1348-1354. doi: 10.1038/ng1896
|
| [57] |
Lieberman-Aiden E, van Berkum NL, Williams L, et al. (2009) Comprehensive mapping of long-range interactions reveals folding principles of the human genome. Science 326: 289-293. doi: 10.1126/science.1181369
|
| [58] |
Bantignies F, Roure V, Comet I, et al. (2011) Polycomb-Dependent Regulatory Contacts between Distant Hox Loci in Drosophila. Cell 144: 214-26. doi: 10.1016/j.cell.2010.12.026
|
| [59] |
Lanzuolo C, Roure V, Dekker J, et al. (2007) Polycomb response elements mediate the formation of chromosome higher-order structures in the bithorax complex. Nat Cell Biol 9: 1167-1174. doi: 10.1038/ncb1637
|
| [60] |
Lo Sardo F, Lanzuolo C, Comoglio F, et al. (2013) PcG-Mediated Higher-Order Chromatin Structures Modulate Replication Programs at the Drosophila BX-C. PLoS Genet 9: e1003283. doi: 10.1371/journal.pgen.1003283
|
| [61] |
Tolhuis B, Blom M, Kerkhoven RM, et al. (2011) Interactions among Polycomb domains are guided by chromosome architecture. PLoS Genet 7: e1001343. doi: 10.1371/journal.pgen.1001343
|
| [62] |
Lanzuolo C, Lo Sardo F, Diamantini A, et al. (2011) PcG Complexes Set the Stage for Epigenetic Inheritance of Gene Silencing in Early S Phase before Replication. PLoS Genet 7: e1002370. doi: 10.1371/journal.pgen.1002370
|
| [63] |
Schoenfelder S, Sugar R, Dimond A, et al. (2015) Polycomb repressive complex PRC1 spatially constrains the mouse embryonic stem cell genome. Nat Genet 47: 1179-1186. doi: 10.1038/ng.3393
|
| [64] |
Ringrose L, Paro R (2007) Polycomb/Trithorax response elements and epigenetic memory of cell identity. Development 134: 223-232. doi: 10.1242/dev.02723
|
| [65] |
Dekker J, Marti-Renom MA, Mirny LA (2013) Exploring the three-dimensional organization of genomes: interpreting chromatin interaction data. Nat Rev Genet 14: 390-403. doi: 10.1038/nrg3454
|
| [66] |
Dixon JR, Selvaraj S, Yue F, et al. (2012) Topological domains in mammalian genomes identified by analysis of chromatin interactions. Nature 485: 376-380. doi: 10.1038/nature11082
|
| [67] |
Hou C, Li L, Qin ZS, et al. (2012) Gene density, transcription, and insulators contribute to the partition of the Drosophila genome into physical domains. Mol Cell 48: 471-484. doi: 10.1016/j.molcel.2012.08.031
|
| [68] |
Andrey G, Montavon T, Mascrez B, et al. (2013) A switch between topological domains underlies HoxD genes collinearity in mouse limbs. Science 340: 1234167. doi: 10.1126/science.1234167
|
| [69] |
Cremer T, Cremer M, Dietzel S, et al. (2006) Chromosome territories--a functional nuclear landscape. Curr Opin Cell Biol 18: 307-316. doi: 10.1016/j.ceb.2006.04.007
|
| [70] |
Parada LA, McQueen PG, Misteli T (2004) Tissue-specific spatial organization of genomes. Genome Biol 5: R44. doi: 10.1186/gb-2004-5-7-r44
|
| [71] |
Tanabe H, Muller S, Neusser M, et al. (2002) Evolutionary conservation of chromosome territory arrangements in cell nuclei from higher primates. Proc Natl Acad Sci U S A 99: 4424-4429. doi: 10.1073/pnas.072618599
|
| [72] |
Meaburn KJ, Misteli T (2007) Cell biology: chromosome territories. Nature 445: 379-781. doi: 10.1038/445379a
|
| [73] | Cremer T, Kreth G, Koester H, et al. (2000) Chromosome territories, interchromatin domain compartment, and nuclear matrix: an integrated view of the functional nuclear architecture. Crit Rev Eukaryot Gene Expr 10: 179-212. |
| [74] |
Schneider R, Grosschedl R (2007) Dynamics and interplay of nuclear architecture, genome organization, and gene expression. Genes Dev 21: 3027-3043. doi: 10.1101/gad.1604607
|
| [75] |
Cremer M, von Hase J, Volm T, et al. (2001) Non-random radial higher-order chromatin arrangements in nuclei of diploid human cells. Chromosome Res 9: 541-567. doi: 10.1023/A:1012495201697
|
| [76] |
Rajapakse I, Groudine M (2011) On emerging nuclear order. J Cell Biol 192: 711-721. doi: 10.1083/jcb.201010129
|
| [77] |
Albiez H, Cremer M, Tiberi C, et al. (2006) Chromatin domains and the interchromatin compartment form structurally defined and functionally interacting nuclear networks. Chromosome Res 14: 707-733. doi: 10.1007/s10577-006-1086-x
|
| [78] |
Mahy NL, Perry PE, Gilchrist S, et al. (2002) Spatial organization of active and inactive genes and noncoding DNA within chromosome territories. J Cell Biol 157: 579-589. doi: 10.1083/jcb.200111071
|
| [79] |
Branco MR, Pombo A (2006) Intermingling of chromosome territories in interphase suggests role in translocations and transcription-dependent associations. PLoS Biol 4: e138. doi: 10.1371/journal.pbio.0040138
|
| [80] |
Meister P, Taddei A (2013) Building silent compartments at the nuclear periphery: a recurrent theme. Curr Opin Genet Dev 23: 96-103. doi: 10.1016/j.gde.2012.12.001
|
| [81] |
Deniaud E, Bickmore WA (2009) Transcription and the nuclear periphery: edge of darkness? Curr Opin Genet Dev 19: 187-191. doi: 10.1016/j.gde.2009.01.005
|
| [82] |
Andrulis ED, Neiman AM, Zappulla DC, et al. (1998) Perinuclear localization of chromatin facilitates transcriptional silencing. Nature 394: 592-595. doi: 10.1038/29100
|
| [83] |
Finlan LE, Sproul D, Thomson I, et al. (2008) Recruitment to the nuclear periphery can alter expression of genes in human cells. PLoS Genet 4: e1000039. doi: 10.1371/journal.pgen.1000039
|
| [84] |
Towbin BD, Gonzalez-Aguilera C, Sack R, et al. (2012) Step-wise methylation of histone H3K9 positions heterochromatin at the nuclear periphery. Cell 150: 934-947. doi: 10.1016/j.cell.2012.06.051
|
| [85] |
Steglich B, Sazer S, Ekwall K (2013) Transcriptional regulation at the yeast nuclear envelope. Nucleus 4: 379-389. doi: 10.4161/nucl.26394
|
| [86] |
Dundr M (2012) Nuclear bodies: multifunctional companions of the genome. Curr Opin Cell Biol 24: 415-422. doi: 10.1016/j.ceb.2012.03.010
|
| [87] |
Zhu L, Brangwynne CP (2015) Nuclear bodies: the emerging biophysics of nucleoplasmic phases. Curr Opin Cell Biol 34: 23-30. doi: 10.1016/j.ceb.2015.04.003
|
| [88] |
Cmarko D, Verschure PJ, Otte AP, et al. (2003) Polycomb group gene silencing proteins are concentrated in the perichromatin compartment of the mammalian nucleus. J Cell Sci 116: 335-343. doi: 10.1242/jcs.00225
|
| [89] |
Isono K, Endo TA, Ku M, et al. (2013) SAM domain polymerization links subnuclear clustering of PRC1 to gene silencing. Dev Cell 26: 565-577. doi: 10.1016/j.devcel.2013.08.016
|
| [90] |
Gonzalez I, Mateos-Langerak J, Thomas A, et al. (2014) Identification of regulators of the three-dimensional polycomb organization by a microscopy-based genome-wide RNAi screen. Mol Cell 54: 485-499. doi: 10.1016/j.molcel.2014.03.004
|
| [91] |
Vandenbunder B, Fourre N, Leray A, et al. (2014) PRC1 components exhibit different binding kinetics in Polycomb bodies. Biol Cell 106: 111-125. doi: 10.1111/boc.201300077
|
| [92] |
Cheutin T, Cavalli G (2012) Progressive Polycomb Assembly on H3K27me3 Compartments Generates Polycomb Bodies with Developmentally Regulated Motion. PLoS Genet 8: e1002465. doi: 10.1371/journal.pgen.1002465
|
| [93] |
Ren X, Vincenz C, Kerppola TK (2008) Changes in the distributions and dynamics of polycomb repressive complexes during embryonic stem cell differentiation. Mol Cell Biol 28: 2884-2895. doi: 10.1128/MCB.00949-07
|
| [94] |
Gao Z, Lee P, Stafford JM, et al. (2014) An AUTS2-Polycomb complex activates gene expression in the CNS. Nature 516: 349-354. doi: 10.1038/nature13921
|
| [95] | Mousavi K, Zare H, Wang AH, et al. (2011) Polycomb Protein Ezh1 Promotes RNA Polymerase II Elongation. Mol Cell. |
| [96] |
Goldman RD, Gruenbaum Y, Moir RD, et al. (2002) Nuclear lamins: building blocks of nuclear architecture. Genes Dev 16: 533-547. doi: 10.1101/gad.960502
|
| [97] |
Goldman AE, Maul G, Steinert PM, et al. (1986) Keratin-like proteins that coisolate with intermediate filaments of BHK-21 cells are nuclear lamins. Proc Natl Acad Sci U S A 83: 3839-3843. doi: 10.1073/pnas.83.11.3839
|
| [98] |
McKeon FD, Kirschner MW, Caput D (1986) Homologies in both primary and secondary structure between nuclear envelope and intermediate filament proteins. Nature 319: 463-468. doi: 10.1038/319463a0
|
| [99] | Rober RA, Sauter H, Weber K, et al. (1990) Cells of the cellular immune and hemopoietic system of the mouse lack lamins A/C: distinction versus other somatic cells. J Cell Sci 95 ( Pt 4): 587-598. |
| [100] |
Solovei I, Wang AS, Thanisch K, et al. (2013) LBR and lamin A/C sequentially tether peripheral heterochromatin and inversely regulate differentiation. Cell 152: 584-598. doi: 10.1016/j.cell.2013.01.009
|
| [101] | Guilly MN, Bensussan A, Bourge JF, et al. (1987) A human T lymphoblastic cell line lacks lamins A and C. Embo J 6: 3795-3799. |
| [102] |
Stewart C, Burke B (1987) Teratocarcinoma stem cells and early mouse embryos contain only a single major lamin polypeptide closely resembling lamin B. Cell 51: 383-392. doi: 10.1016/0092-8674(87)90634-9
|
| [103] |
Kolb T, Maass K, Hergt M, et al. (2011) Lamin A and lamin C form homodimers and coexist in higher complex forms both in the nucleoplasmic fraction and in the lamina of cultured human cells. Nucleus 2: 425-433. doi: 10.4161/nucl.2.5.17765
|
| [104] |
Shimi T, Pfleghaar K, Kojima S, et al. (2008) The A- and B-type nuclear lamin networks: microdomains involved in chromatin organization and transcription. Genes Dev 22: 3409-3421. doi: 10.1101/gad.1735208
|
| [105] |
Dubinska-Magiera M, Zaremba-Czogalla M, Rzepecki R (2013) Muscle development, regeneration and laminopathies: how lamins or lamina-associated proteins can contribute to muscle development, regeneration and disease. Cell Mol Life Sci 70: 2713-2741. doi: 10.1007/s00018-012-1190-3
|
| [106] |
Collas P, Lund EG, Oldenburg AR (2014) Closing the (nuclear) envelope on the genome: How nuclear lamins interact with promoters and modulate gene expression. Bioessays 36: 75-83. doi: 10.1002/bies.201300138
|
| [107] |
Meuleman W, Peric-Hupkes D, Kind J, et al. (2013) Constitutive nuclear lamina-genome interactions are highly conserved and associated with A/T-rich sequence. Genome Res 23: 270-280. doi: 10.1101/gr.141028.112
|
| [108] |
Kind J, Pagie L, Ortabozkoyun H, et al. (2013) Single-cell dynamics of genome-nuclear lamina interactions. Cell 153: 178-192. doi: 10.1016/j.cell.2013.02.028
|
| [109] |
Kind J, van Steensel B (2010) Genome-nuclear lamina interactions and gene regulation. Curr Opin Cell Biol 22: 320-325. doi: 10.1016/j.ceb.2010.04.002
|
| [110] | Lund E, Oldenburg A, Delbarre E, et al. (2013) Lamin A/C-promoter interactions specify chromatin state-dependent transcription outcomes. Genome Res. |
| [111] |
Pickersgill H, Kalverda B, de Wit E, et al. (2006) Characterization of the Drosophila melanogaster genome at the nuclear lamina. Nat Genet 38: 1005-1014. doi: 10.1038/ng1852
|
| [112] |
Guelen L, Pagie L, Brasset E, et al. (2008) Domain organization of human chromosomes revealed by mapping of nuclear lamina interactions. Nature 453: 948-951. doi: 10.1038/nature06947
|
| [113] |
Kind J, Pagie L, de Vries SS, et al. (2015) Genome-wide Maps of Nuclear Lamina Interactions in Single Human Cells. Cell 163: 134-147. doi: 10.1016/j.cell.2015.08.040
|
| [114] | Mattout A, Pike BL, Towbin BD, et al. (2011) An EDMD mutation in C. elegans lamin blocks muscle-specific gene relocation and compromises muscle integrity. Curr Biol 21: 1603-1614. |
| [115] |
Peric-Hupkes D, Meuleman W, Pagie L, et al. (2010) Molecular maps of the reorganization of genome-nuclear lamina interactions during differentiation. Mol Cell 38: 603-613. doi: 10.1016/j.molcel.2010.03.016
|
| [116] |
Zaidi SK, Young DW, Montecino MA, et al. (2010) Mitotic bookmarking of genes: a novel dimension to epigenetic control. Nat Rev Genet 11: 583-589. doi: 10.1038/nrg2827
|
| [117] |
Kind J, van Steensel B (2014) Stochastic genome-nuclear lamina interactions: Modulating roles of Lamin A and BAF. Nucleus 5: 124-130. doi: 10.4161/nucl.28825
|
| [118] |
Amendola M, van Steensel B (2015) Nuclear lamins are not required for lamina-associated domain organization in mouse embryonic stem cells. EMBO Rep 16: 610-617. doi: 10.15252/embr.201439789
|
| [119] |
Osmanagic-Myers S, Dechat T, Foisner R (2015) Lamins at the crossroads of mechanosignaling. Genes Dev 29: 225-237. doi: 10.1101/gad.255968.114
|
| [120] |
Haque F, Lloyd DJ, Smallwood DT, et al. (2006) SUN1 interacts with nuclear lamin A and cytoplasmic nesprins to provide a physical connection between the nuclear lamina and the cytoskeleton. Mol Cell Biol 26: 3738-3751. doi: 10.1128/MCB.26.10.3738-3751.2006
|
| [121] | Gruenbaum Y, Foisner R (2015) Lamins: Nuclear Intermediate Filament Proteins with Fundamental Functions in Nuclear Mechanics and Genome Regulation. Annu Rev Biochem. |
| [122] |
Swift J, Ivanovska IL, Buxboim A, et al. (2013) Nuclear lamin-A scales with tissue stiffness and enhances matrix-directed differentiation. Science 341: 1240104. doi: 10.1126/science.1240104
|
| [123] |
Padiath QS, Saigoh K, Schiffmann R, et al. (2006) Lamin B1 duplications cause autosomal dominant leukodystrophy. Nat Genet 38: 1114-1123. doi: 10.1038/ng1872
|
| [124] |
Hegele RA, Cao H, Liu DM, et al. (2006) Sequencing of the reannotated LMNB2 gene reveals novel mutations in patients with acquired partial lipodystrophy. Am J Hum Genet 79: 383-389. doi: 10.1086/505885
|
| [125] | Harborth J, Elbashir SM, Bechert K, et al. (2001) Identification of essential genes in cultured mammalian cells using small interfering RNAs. J Cell Sci 114: 4557-4565. |
| [126] | Zaremba-Czogalla M, Dubinska-Magiera M, Rzepecki R (2011) Laminopathies: the molecular background of the disease and the prospects for its treatment. Cell Mol Biol Lett 16: 114-148. |
| [127] |
Broers JL, Peeters EA, Kuijpers HJ, et al. (2004) Decreased mechanical stiffness in LMNA-/- cells is caused by defective nucleo-cytoskeletal integrity: implications for the development of laminopathies. Hum Mol Genet 13: 2567-2580. doi: 10.1093/hmg/ddh295
|
| [128] |
Sullivan T, Escalante-Alcalde D, Bhatt H, et al. (1999) Loss of A-type lamin expression compromises nuclear envelope integrity leading to muscular dystrophy. J Cell Biol 147: 913-920. doi: 10.1083/jcb.147.5.913
|
| [129] |
Hernandez L, Roux KJ, Wong ES, et al. (2010) Functional coupling between the extracellular matrix and nuclear lamina by Wnt signaling in progeria. Dev Cell 19: 413-425. doi: 10.1016/j.devcel.2010.08.013
|
| [130] | Sinha JK, Ghosh S, Raghunath M (2014) Progeria: a rare genetic premature ageing disorder. Indian J Med Res 139: 667-674. |
| [131] |
McCord RP, Nazario-Toole A, Zhang H, et al. (2013) Correlated alterations in genome organization, histone methylation, and DNA-lamin A/C interactions in Hutchinson-Gilford progeria syndrome. Genome Res 23: 260-269. doi: 10.1101/gr.138032.112
|
| [132] |
Shumaker DK, Dechat T, Kohlmaier A, et al. (2006) Mutant nuclear lamin A leads to progressive alterations of epigenetic control in premature aging. Proc Natl Acad Sci U S A 103: 8703-8708. doi: 10.1073/pnas.0602569103
|
| [133] |
Landires I, Pascale JM, Motta J (2007) The position of the mutation within the LMNA gene determines the type and extent of tissue involvement in laminopathies. Clin Genet 71: 592-593; author reply 594-596. doi: 10.1111/j.1399-0004.2007.00772.x
|
| [134] |
Scharner J, Gnocchi VF, Ellis JA, et al. (2010) Genotype-phenotype correlations in laminopathies: how does fate translate? Biochem Soc Trans 38: 257-262. doi: 10.1042/BST0380257
|
| [135] |
Sewry CA, Brown SC, Mercuri E, et al. (2001) Skeletal muscle pathology in autosomal dominant Emery-Dreifuss muscular dystrophy with lamin A/C mutations. Neuropathol Appl Neurobiol 27: 281-290. doi: 10.1046/j.0305-1846.2001.00323.x
|
| [136] | Cesarini E, Mozzetta C, Marullo F, et al. (2015) Lamin A/C sustains PcG proteins architecture maintaining transcriptional repression at target genes. J Cell Biol. |
| [137] |
Ptak C, Aitchison JD, Wozniak RW (2014) The multifunctional nuclear pore complex: a platform for controlling gene expression. Curr Opin Cell Biol 28: 46-53. doi: 10.1016/j.ceb.2014.02.001
|
| [138] |
Blobel G (1985) Gene gating: a hypothesis. Proc Natl Acad Sci U S A 82: 8527-8529. doi: 10.1073/pnas.82.24.8527
|
| [139] |
Cabal GG, Genovesio A, Rodriguez-Navarro S, et al. (2006) SAGA interacting factors confine sub-diffusion of transcribed genes to the nuclear envelope. Nature 441: 770-773. doi: 10.1038/nature04752
|
| [140] |
Taddei A, Van Houwe G, Hediger F, et al. (2006) Nuclear pore association confers optimal expression levels for an inducible yeast gene. Nature 441: 774-778. doi: 10.1038/nature04845
|
| [141] |
Tan-Wong SM, Wijayatilake HD, Proudfoot NJ (2009) Gene loops function to maintain transcriptional memory through interaction with the nuclear pore complex. Genes Dev 23: 2610-2624. doi: 10.1101/gad.1823209
|
| [142] |
Green EM, Jiang Y, Joyner R, et al. (2012) A negative feedback loop at the nuclear periphery regulates GAL gene expression. Mol Biol Cell 23: 1367-1375. doi: 10.1091/mbc.E11-06-0547
|
| [143] | Yoshida T, Shimada K, Oma Y, et al. (2010) Actin-related protein Arp6 influences H2A.Z-dependent and -independent gene expression and links ribosomal protein genes to nuclear pores. PLoS Genet 6: e1000910. |
| [144] |
Van de Vosse DW, Wan Y, Lapetina DL, et al. (2013) A role for the nucleoporin Nup170p in chromatin structure and gene silencing. Cell 152: 969-983. doi: 10.1016/j.cell.2013.01.049
|
| [145] |
Galy V, Olivo-Marin JC, Scherthan H, et al. (2000) Nuclear pore complexes in the organization of silent telomeric chromatin. Nature 403: 108-112. doi: 10.1038/47528
|
| [146] |
Buchwalter AL, Liang Y, Hetzer MW (2014) Nup50 is required for cell differentiation and exhibits transcription-dependent dynamics. Mol Biol Cell 25: 2472-2484. doi: 10.1091/mbc.E14-04-0865
|
| [147] |
Gomez-Cavazos JS, Hetzer MW (2015) The nucleoporin gp210/Nup210 controls muscle differentiation by regulating nuclear envelope/ER homeostasis. J Cell Biol 208: 671-681. doi: 10.1083/jcb.201410047
|
| [148] |
Iwamoto M, Koujin T, Osakada H, et al. (2015) Biased assembly of the nuclear pore complex is required for somatic and germline nuclear differentiation in Tetrahymena. J Cell Sci 128: 1812-1823. doi: 10.1242/jcs.167353
|
| [149] |
Liang Y, Franks TM, Marchetto MC, et al. (2013) Dynamic association of NUP98 with the human genome. PLoS Genet 9: e1003308. doi: 10.1371/journal.pgen.1003308
|
| [150] |
Lupu F, Alves A, Anderson K, et al. (2008) Nuclear pore composition regulates neural stem/progenitor cell differentiation in the mouse embryo. Dev Cell 14: 831-842. doi: 10.1016/j.devcel.2008.03.011
|
| [151] |
D'Angelo MA, Gomez-Cavazos JS, Mei A, et al. (2012) A change in nuclear pore complex composition regulates cell differentiation. Dev Cell 22: 446-458. doi: 10.1016/j.devcel.2011.11.021
|
| [152] |
Lin Y, Yang Y, Li W, et al. (2012) Reciprocal regulation of Akt and Oct4 promotes the self-renewal and survival of embryonal carcinoma cells. Mol Cell 48: 627-640. doi: 10.1016/j.molcel.2012.08.030
|
| [153] | Yang J, Cai N, Yi F, et al. (2014) Gating pluripotency via nuclear pores. Trends Mol Med 20: 1-7. |
| [154] |
Zhang X, Chen S, Yoo S, et al. (2008) Mutation in nuclear pore component NUP155 leads to atrial fibrillation and early sudden cardiac death. Cell 135: 1017-1027. doi: 10.1016/j.cell.2008.10.022
|
| [155] |
Basel-Vanagaite L, Muncher L, Straussberg R, et al. (2006) Mutated nup62 causes autosomal recessive infantile bilateral striatal necrosis. Ann Neurol 60: 214-222. doi: 10.1002/ana.20902
|
| [156] |
van Koningsbruggen S, Gierlinski M, Schofield P, et al. (2010) High-resolution whole-genome sequencing reveals that specific chromatin domains from most human chromosomes associate with nucleoli. Mol Biol Cell 21: 3735-3748. doi: 10.1091/mbc.E10-06-0508
|
| [157] |
Nemeth A, Conesa A, Santoyo-Lopez J, et al. (2010) Initial genomics of the human nucleolus. PLoS Genet 6: e1000889. doi: 10.1371/journal.pgen.1000889
|
| [158] |
Kim SK, Lee H, Han K, et al. (2014) SET7/9 methylation of the pluripotency factor LIN28A is a nucleolar localization mechanism that blocks let-7 biogenesis in human ESCs. Cell Stem Cell 15: 735-749. doi: 10.1016/j.stem.2014.10.016
|
| [159] |
Savic N, Bar D, Leone S, et al. (2014) lncRNA maturation to initiate heterochromatin formation in the nucleolus is required for exit from pluripotency in ESCs. Cell Stem Cell 15: 720-734. doi: 10.1016/j.stem.2014.10.005
|
| [160] | Talamas JA, Capelson M (2015) Nuclear envelope and genome interactions in cell fate. Front Genet 6: 95. |
| [161] |
Fischer AH (2014) The diagnostic pathology of the nuclear envelope in human cancers. Adv Exp Med Biol 773: 49-75. doi: 10.1007/978-1-4899-8032-8_3
|
| [162] |
de Las Heras JI, Schirmer EC (2014) The nuclear envelope and cancer: a diagnostic perspective and historical overview. Adv Exp Med Biol 773: 5-26. doi: 10.1007/978-1-4899-8032-8_1
|
| [163] |
Skvortsov S, Schafer G, Stasyk T, et al. (2011) Proteomics profiling of microdissected low- and high-grade prostate tumors identifies Lamin A as a discriminatory biomarker. J Proteome Res 10: 259-268. doi: 10.1021/pr100921j
|
| [164] |
Willis ND, Cox TR, Rahman-Casans SF, et al. (2008) Lamin A/C is a risk biomarker in colorectal cancer. PLoS One 3: e2988. doi: 10.1371/journal.pone.0002988
|
| [165] |
Vargas JD, Hatch EM, Anderson DJ, et al. (2012) Transient nuclear envelope rupturing during interphase in human cancer cells. Nucleus 3: 88-100. doi: 10.4161/nucl.18954
|
| [166] |
Fischer AH, Bardarov S, Jr., Jiang Z (2004) Molecular aspects of diagnostic nucleolar and nuclear envelope changes in prostate cancer. J Cell Biochem 91: 170-184. doi: 10.1002/jcb.10735
|
| [167] |
Suresh S (2007) Biomechanics and biophysics of cancer cells. Acta Biomater 3: 413-438. doi: 10.1016/j.actbio.2007.04.002
|
| [168] |
Lammerding J, Fong LG, Ji JY, et al. (2006) Lamins A and C but not lamin B1 regulate nuclear mechanics. J Biol Chem 281: 25768-25780. doi: 10.1074/jbc.M513511200
|
| [169] | Chow KH, Factor RE, Ullman KS (2012) The nuclear envelope environment and its cancer connections. Nat Rev Cancer 12: 196-209. |
| [170] |
Mjelle R, Hegre SA, Aas PA, et al. (2015) Cell cycle regulation of human DNA repair and chromatin remodeling genes. DNA Repair (Amst) 30: 53-67. doi: 10.1016/j.dnarep.2015.03.007
|
| [171] |
Yi C, He C (2013) DNA repair by reversal of DNA damage. Cold Spring Harb Perspect Biol 5: a012575. doi: 10.1101/cshperspect.a012575
|
| [172] | Davis AJ, Chen DJ (2013) DNA double strand break repair via non-homologous end-joining. Transl Cancer Res 2: 130-143. |
| [173] |
Shroff R, Arbel-Eden A, Pilch D, et al. (2004) Distribution and dynamics of chromatin modification induced by a defined DNA double-strand break. Curr Biol 14: 1703-1711. doi: 10.1016/j.cub.2004.09.047
|
| [174] |
Jazayeri A, Falck J, Lukas C, et al. (2006) ATM- and cell cycle-dependent regulation of ATR in response to DNA double-strand breaks. Nat Cell Biol 8: 37-45. doi: 10.1038/ncb1337
|
| [175] |
Radhakrishnan SK, Jette N, Lees-Miller SP (2014) Non-homologous end joining: emerging themes and unanswered questions. DNA Repair (Amst) 17: 2-8. doi: 10.1016/j.dnarep.2014.01.009
|
| [176] |
Shrivastav M, De Haro LP, Nickoloff JA (2008) Regulation of DNA double-strand break repair pathway choice. Cell Res 18: 134-147. doi: 10.1038/cr.2007.111
|
| [177] |
Lemaitre C, Grabarz A, Tsouroula K, et al. (2014) Nuclear position dictates DNA repair pathway choice. Genes Dev 28: 2450-2463. doi: 10.1101/gad.248369.114
|
| [178] |
Aymard F, Bugler B, Schmidt CK, et al. (2014) Transcriptionally active chromatin recruits homologous recombination at DNA double-strand breaks. Nat Struct Mol Biol 21: 366-374. doi: 10.1038/nsmb.2796
|
| [179] |
Horigome C, Oma Y, Konishi T, et al. (2014) SWR1 and INO80 chromatin remodelers contribute to DNA double-strand break perinuclear anchorage site choice. Mol Cell 55: 626-639. doi: 10.1016/j.molcel.2014.06.027
|
| [180] |
Dion V, Gasser SM (2013) Chromatin movement in the maintenance of genome stability. Cell 152: 1355-1364. doi: 10.1016/j.cell.2013.02.010
|
| [181] |
Dion V, Kalck V, Horigome C, et al. (2012) Increased mobility of double-strand breaks requires Mec1, Rad9 and the homologous recombination machinery. Nat Cell Biol 14: 502-509. doi: 10.1038/ncb2465
|
| [182] |
Seeber A, Dion V, Gasser SM (2013) Checkpoint kinases and the INO80 nucleosome remodeling complex enhance global chromatin mobility in response to DNA damage. Genes Dev 27: 1999-2008. doi: 10.1101/gad.222992.113
|
| [183] |
Neumann FR, Dion V, Gehlen LR, et al. (2012) Targeted INO80 enhances subnuclear chromatin movement and ectopic homologous recombination. Genes Dev 26: 369-383. doi: 10.1101/gad.176156.111
|
| [184] |
Mahen R, Hattori H, Lee M, et al. (2013) A-type lamins maintain the positional stability of DNA damage repair foci in mammalian nuclei. PLoS One 8: e61893. doi: 10.1371/journal.pone.0061893
|
| [185] |
van Sluis M, McStay B (2015) A localized nucleolar DNA damage response facilitates recruitment of the homology-directed repair machinery independent of cell cycle stage. Genes Dev 29: 1151-1163. doi: 10.1101/gad.260703.115
|
| [186] |
Tang J, Cho NW, Cui G, et al. (2013) Acetylation limits 53BP1 association with damaged chromatin to promote homologous recombination. Nat Struct Mol Biol 20: 317-325. doi: 10.1038/nsmb.2499
|
| [187] |
Botuyan MV, Lee J, Ward IM, et al. (2006) Structural basis for the methylation state-specific recognition of histone H4-K20 by 53BP1 and Crb2 in DNA repair. Cell 127: 1361-1373. doi: 10.1016/j.cell.2006.10.043
|
| [188] | Misteli T, Soutoglou E (2009) The emerging role of nuclear architecture in DNA repair and genome maintenance. Nat Rev Mol Cell Biol 10: 243-254. |
| [189] |
Constantinescu D, Csoka AB, Navara CS, et al. (2010) Defective DSB repair correlates with abnormal nuclear morphology and is improved with FTI treatment in Hutchinson-Gilford progeria syndrome fibroblasts. Exp Cell Res 316: 2747-2759. doi: 10.1016/j.yexcr.2010.05.015
|
| [190] |
Lattanzi G, Marmiroli S, Facchini A, et al. (2012) Nuclear damages and oxidative stress: new perspectives for laminopathies. Eur J Histochem 56: e45. doi: 10.4081/ejh.2012.e45
|
| [191] |
Richards SA, Muter J, Ritchie P, et al. (2011) The accumulation of un-repairable DNA damage in laminopathy progeria fibroblasts is caused by ROS generation and is prevented by treatment with N-acetyl cysteine. Hum Mol Genet 20: 3997-4004. doi: 10.1093/hmg/ddr327
|
| [192] |
Allen RG, Tresini M (2000) Oxidative stress and gene regulation. Free Radic Biol Med 28: 463-499. doi: 10.1016/S0891-5849(99)00242-7
|
| [193] |
Sieprath T, Corne T, Nooteboom M, et al. (2015) Sustained accumulation of prelamin A and depletion of lamin A/C both cause oxidative stress and mitochondrial dysfunction but induce different cell fates. Nucleus 6: 236-246. doi: 10.1080/19491034.2015.1050568
|
| [194] |
Pekovic V, Gibbs-Seymour I, Markiewicz E, et al. (2011) Conserved cysteine residues in the mammalian lamin A tail are essential for cellular responses to ROS generation. Aging Cell 10: 1067–1079. doi: 10.1111/j.1474-9726.2011.00750.x
|
| [195] |
Viteri G, Chung YW, Stadtman ER (2010) Effect of progerin on the accumulation of oxidized proteins in fibroblasts from Hutchinson Gilford progeria patients. Mech Ageing Dev 131: 2–8. doi: 10.1016/j.mad.2009.11.006
|
| [196] |
Csoka AB, Cao H, Sammak PJ, et al. (2004) Novel lamin A/C gene (LMNA) mutations in atypical progeroid syndromes. J Med Genet 41: 304–308. doi: 10.1136/jmg.2003.015651
|
| [197] |
Doubaj Y, De Sandre-Giovannoli A, Vera EV, et al. (2012) An inherited LMNA gene mutation in atypical Progeria syndrome. Am J Med Genet A 158A: 2881–2887. doi: 10.1002/ajmg.a.35557
|
| [198] | Seco-Cervera M, Spis M, Garcia-Gimenez JL, et al. (2014) Oxidative stress and antioxidant response in fibroblasts from Werner and atypical Werner syndromes. Aging (Albany NY) 6: 231–245. |
| [199] |
Schroder AR, Shinn P, Chen H, et al. (2002) HIV-1 integration in the human genome favors active genes and local hotspots. Cell 110: 521–529. doi: 10.1016/S0092-8674(02)00864-4
|
| [200] | Wong RW, Mamede JI, Hope TJ (2015) The Impact of Nucleoporin Mediated Chromatin localization and Nuclear Architecture on HIV Integration Site Selection. J Virol. |
| [201] |
Marini B, Kertesz-Farkas A, Ali H, et al. (2015) Nuclear architecture dictates HIV-1 integration site selection. Nature 521: 227–231.202. Lelek M, Casartelli N, Pellin D, et al. (2015) Chromatin organization at the nuclear pore favours HIV replication. Nat Commun 6: 6483. doi: 10.1038/nature14226
|
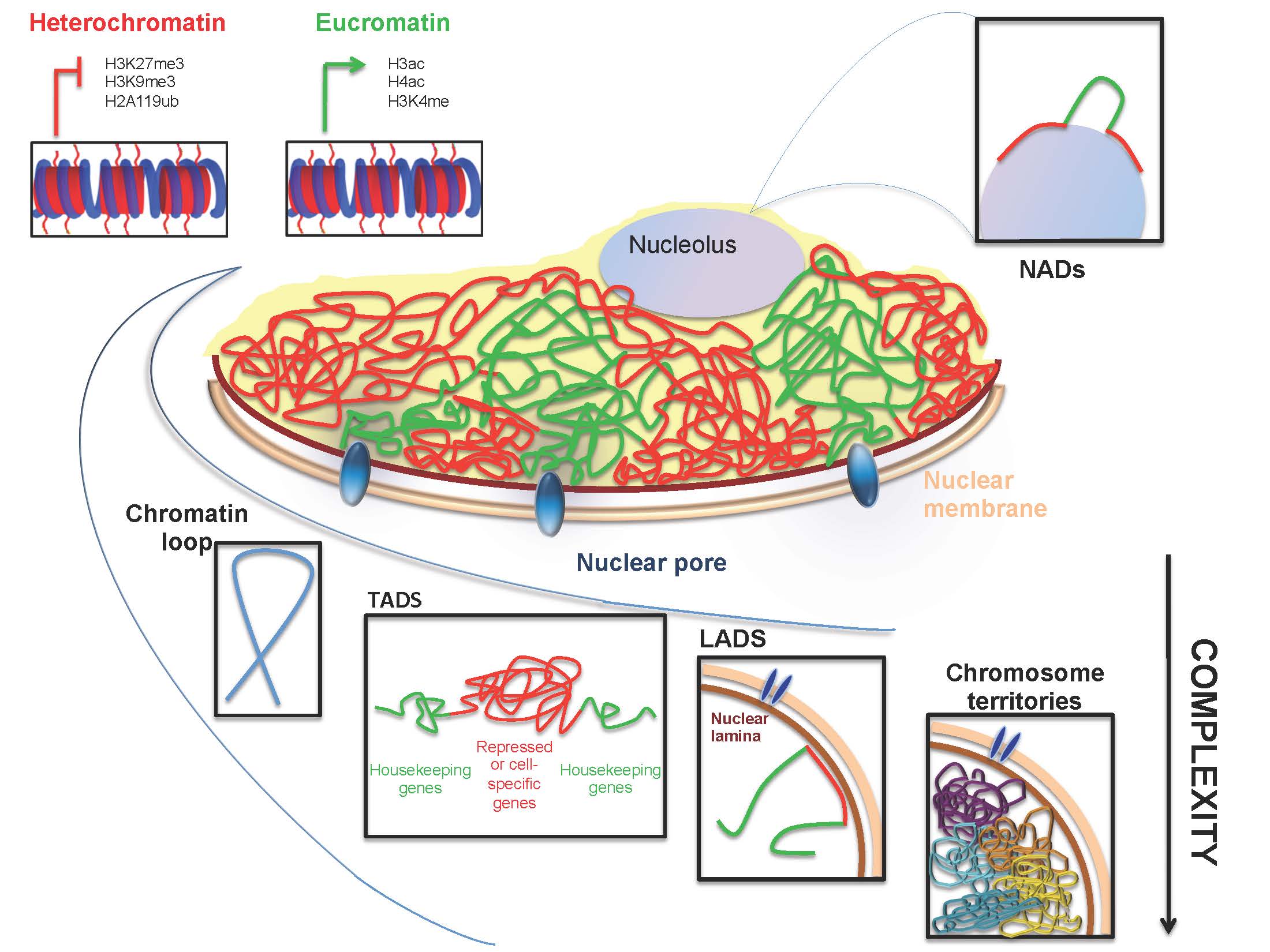
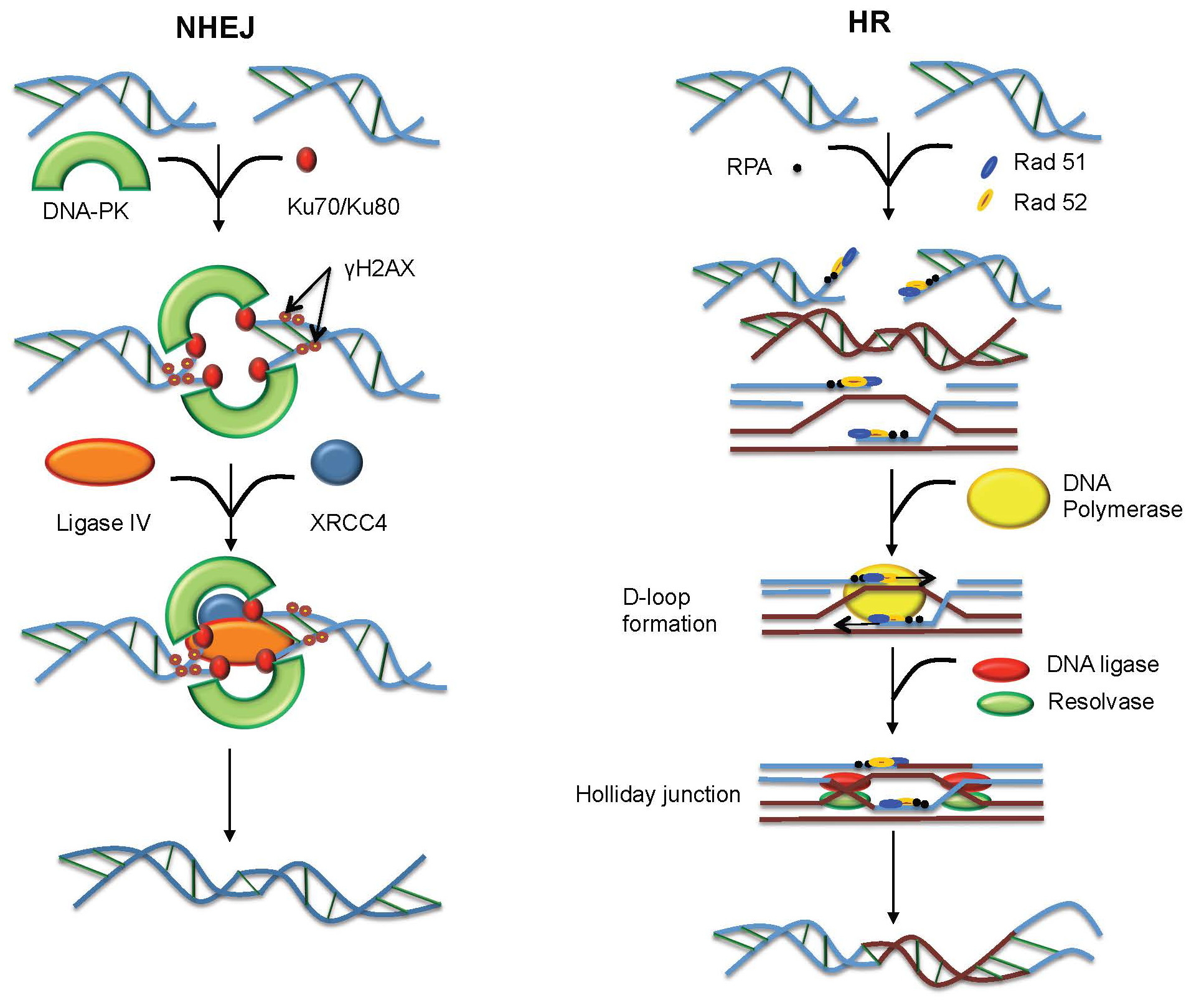
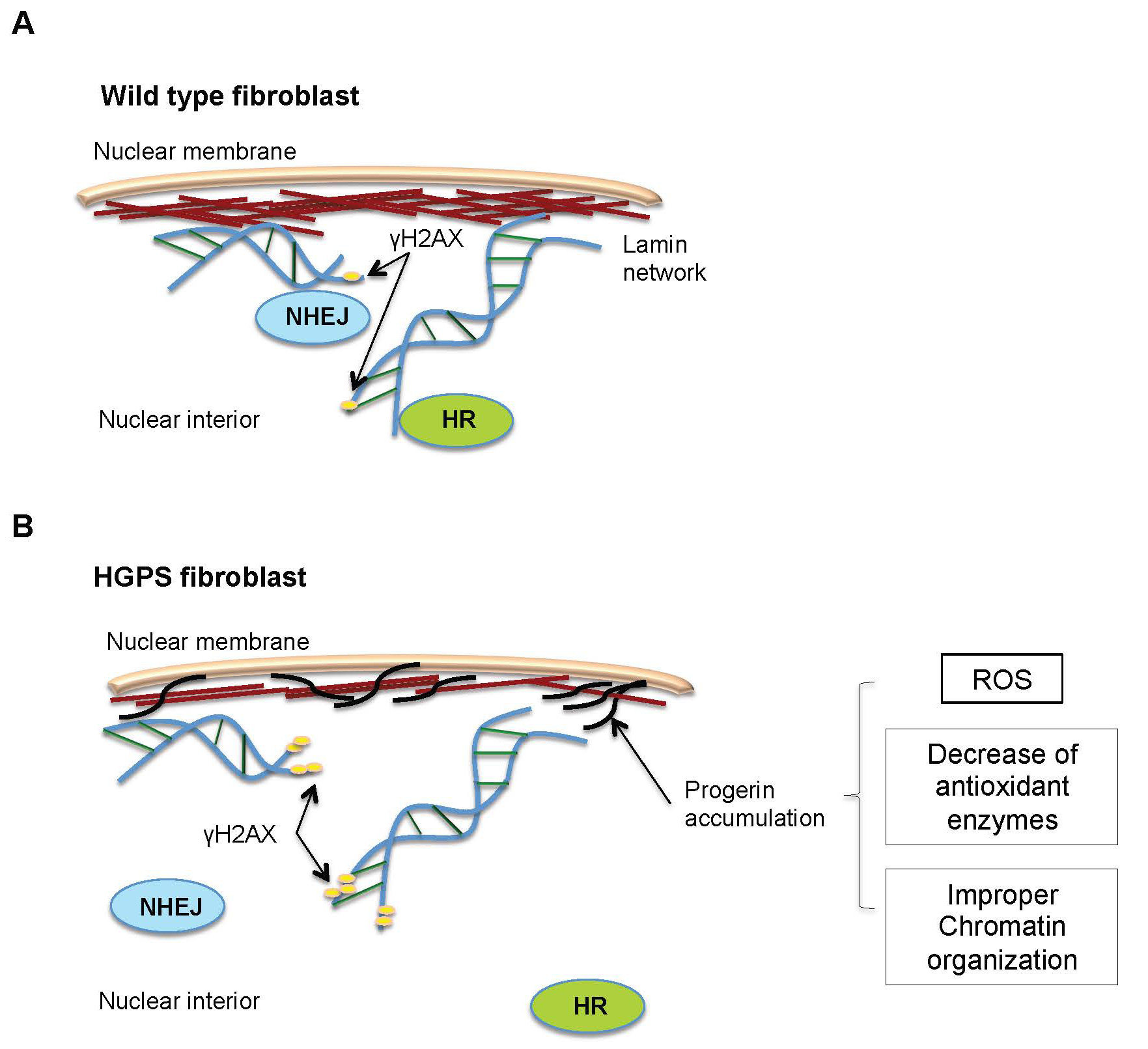
Figures(4) / Tables(1)

Andrea Bianchi, Chiara Lanzuolo. Into the chromatin world: Role of nuclear architecture in epigenome regulation[J]. AIMS Biophysics, 2015, 2(4): 585-612. doi: 10.3934/biophy.2015.4.585







 DownLoad:
DownLoad: