Citation: Keith E. Whitener Jr. Rapid synthesis of thin amorphous carbon films by sugar dehydration and dispersion[J]. AIMS Materials Science, 2016, 3(4): 1309-1320. doi: 10.3934/matersci.2016.4.1309

| [1] | Tro NJ (2011) Chemistry: A Molecular Approach, 2nd ed., Pearson Prentice Hall: Upper Saddle River, NJ, 1013. |
| [2] | Zhang J, Shen W, Pan D, et al. (2010) Controlled synthesis of green and blue luminescent carbon nanoparticles with high yields by the carbonization of sucrose. New J Chem 34: 591–593. |
| [3] | Peng H, Travas-Sejdic J (2009) Simple aqueous solution route to luminescent carbogenic dots from carbohydrates. Chem Mater 21: 5563–5565. |
| [4] | Li H, Kang Z, Liu Y, et al. (2012) Carbon nanodots: synthesis, properties and applications. J Mater Chem 22: 24230–24253. |
| [5] | Pan F, Jin J, Fu X, et al. (2013) Advanced oxygen reduction electrocatalyst based on nitrogen-doped graphene derived from edible sugar and urea. ACS Appl Mater Inter 5: 11108–11114. |
| [6] | Gupta SS, Sreeprasad TS, Maliyekkal SM, et al. (2012) Graphene from sugar and its application in water purification. ACS Appl Mater Inter 4: 4156–4163. |
| [7] | Ruiz-Hitzky E, Darder M, Fernandes FM, et al. (2011) Supported graphene from natural resources: Easy preparation and applications. Adv Mater 23: 5250–5255. |
| [8] | Ruan G, Sun Z, Peng Z, et al. (2011) Growth of graphene from food, insects, and waste. ACS Nano 5: 7601–7607. |
| [9] | Kubo S, White RJ, Yoshizawa N, et al. (2011) Ordered carbohydrate-derived porous carbons. Chem Mater 23: 4882–4885. |
| [10] | Yu C, Fan J, Tian B, et al. (2002) High-yield synthesis of periodic mesoporous silica rods and their replication to mesoporous carbon rods. Adv Mater 14: 1742–1745. |
| [11] | Titirici MM, Antonietti M (2010) Chemistry and materials options of sustainable carbon materials made by hydrothermal carbonization. Chem Soc Rev 39: 103–116. |
| [12] |
Javid A, Kumar M, Yoon S, et al. (2016) Role of surface-electrical properties on the cell-viability of carbon thin films grown in nanodomain morphology. J Phys D Appl Phys 49: 264001. doi: 10.1088/0022-3727/49/26/264001

|
| [13] | Javid A, Kumar M, Han JG (2015) Nanoscale surface conductivity analysis of plasma sputtered carbon thin films. RSC Adv 5: 96360–96365. |
| [14] | Kumar M, Piao JX, Jin SB, et al. (2016) Low temperature plasma processing for cell growth inspired carbon thin films fabrication. Arch Biochem Biophys 605: 41–48. |
| [15] | Piao JX, Kumar M, Javid A, et al. (2016) Pulsed DC-plasma sputtering induced synthesis of hydrogenated carbon thin films for L-929 cell cultivation. Surf Coat Tech. |
| [16] | Ryoo R, Joo SH, Jun S (1999) Synthesis of highly ordered carbon molecular sieves via template-mediated structural transformation. J Phys Chem B 103: 7743–7746. |
| [17] | Su C, Loh KP (2012) Carbocatalysts: Graphene oxide and its derivatives. Acc Chem Res 46: 2275–2285. |
| [18] | Qu L, Liu Y, Baek JB, et al. (2010) Nitrogen-doped graphene as efficient metal-free electrocatalyst for oxygen reduction in fuel cells. ACS Nano 4: 1321–1326. |
| [19] | Vijapur SH, Wang D, Botte GG (2013) The growth of transparent amorphous carbon thin films from coal. Carbon 54: 22–28. |
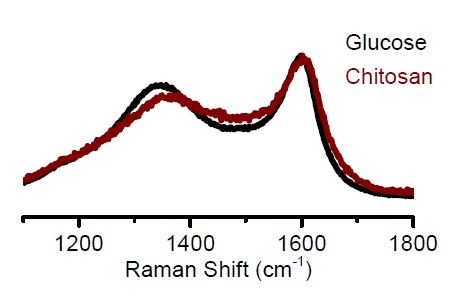
| [20] | Ferrari AC, Robertson J (2001) Origin of the 1150 cm-1 Raman mode in nanocrystalline diamond. Phys Rev B 63: 121405. |
| [21] | Jorio A, Dresselhaus M, Saito R, et al. (2011) Raman Spectroscopy in Graphene Related Systems, Wiley-VCH: Weinheim, Germany. |
| [22] | Kaplas T, Kuzhir P (2016) Ultra-thin graphitic film: Synthesis and physical properties. Nanoscale Res Lett 11: 1–6. |
| [23] | Kaplas T, Svirko Y (2012) Direct deposition of semitransparent conducting pyrolytic carbon films. J Nanophotonics 6: 061703–061703. |
| [24] | Ferrari AC, Basko DM (2013) Raman spectroscopy as a versatile tool for studying the properties of graphene. Nat Nanotechnol 8: 235–246. |
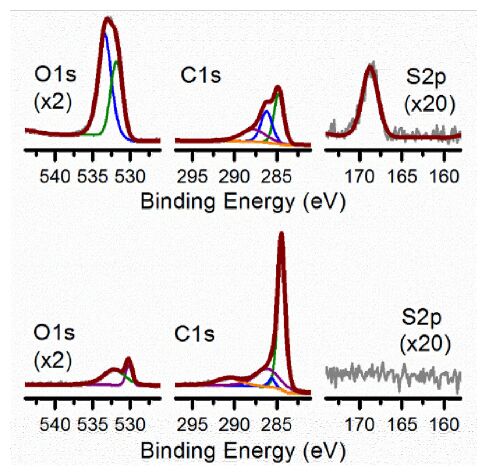
| [25] | Zhou JH, Sui ZJ, Zhu J, et al. (2007) Characterization of surface oxygen complexes on carbon nanofibers by TPD, XPS and FT-IR. Carbon 45: 785–796. |
| [26] | Desimoni E, Casella GI, Morone A, et al. (1990) XPS determination of oxygen-containing functional groups on carbon-fibre surfaces and the cleaning of these surfaces. Surf Interface Anal 15: 627–634. |
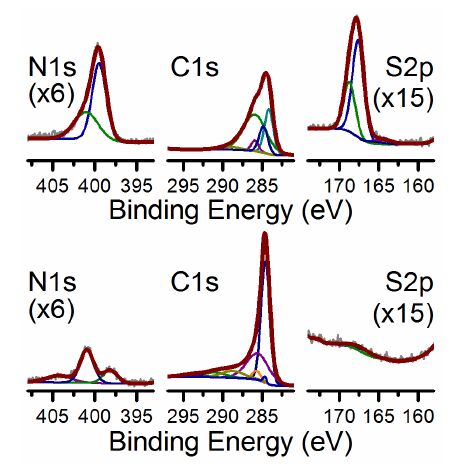
| [27] | López GP, Castner DG, Ratner BD (1991) XPS O1s binding energies for polymers containing hydroxyl, ether, ketone and ester groups. Surf Interface Anal 17: 267–272. |
| [28] | Plomp AJ, Su DS, Jong KPD, et al. (2009) On the nature of oxygen-containing surface groups on carbon nanofibers and their role for Platinum deposition—An XPS and titration study. J Phys Chem C 113: 9865–9869. |
| [29] | Wang H, Maiyalagan T, Wang X (2012) Review on recent progress in nitrogen-doped graphene: Synthesis, characterization, and its potential applications. ACS Catal 2: 781–794. |
| [30] | Shao Y, Zhang S, Engelhard MH, et al. (2010) Nitrogen-doped graphene and its electrochemical applications. J Mater Chem 20: 7491–7496. |
| [31] |
Kondo T, Casolo S, Suzuki T, et al. (2012) Atomic-scale characterization of nitrogen-doped graphite: Effects of dopant nitrogen on the local electronic structure of the surrounding carbon atoms. Phys Rev B 86: 035436. doi: 10.1103/PhysRevB.86.035436
|
| [32] | Lahaye J, Nanse G, Bagreev A, et al. (1999) Porous structure and surface chemistry of nitrogen containing carbons from polymers. Carbon 37: 585–590. |
| [33] | Choudhary V, Burnett RI, Vlachos DG, et al. (2012) Dehydration of Glucose to 5-(Hydroxymethyl)furfural and Anhydroglucose: Thermodynamic Insights. J Phys Chem C 116: 5116–5120. |
| [34] | Franklin RE (1951) Crystallite Growth in Graphitizing and Non-Graphitizing Carbons. Proc R Soc Lond A Math Phys Sci 209: 196–218. |
Figures(7)

Keith E. Whitener Jr. Rapid synthesis of thin amorphous carbon films by sugar dehydration and dispersion[J]. AIMS Materials Science, 2016, 3(4): 1309-1320. doi: 10.3934/matersci.2016.4.1309







 DownLoad:
DownLoad: