Citation: Tara P. Hurst, Caroline Coleman-Vaughan, Indu Patwal, Justin V. McCarthy. Regulated intramembrane proteolysis, innate immunity and therapeutic targets in Alzheimer’s disease[J]. AIMS Molecular Science, 2016, 3(2): 138-157. doi: 10.3934/molsci.2016.2.138

| [1] |
De Strooper B, Saftig P, Craessaerts K, et al. (1998) Deficiency of presenilin-1 inhibits the normal cleavage of amyloid precursor protein. Nature 391: 387-390. doi: 10.1038/34910

|
| [2] |
Jankowsky JL, Fadale DJ, Anderson J, et al. (2004) Mutant presenilins specifically elevate the levels of the 42 residue beta-amyloid peptide in vivo: evidence for augmentation of a 42-specific gamma secretase. Hum Mol Genet 13: 159-170. doi: 10.1093/hmg/ddh019
|
| [3] |
Clark RF, Hutton M, Fuldner M, et al. (1995) The structure of the presenilin 1 (S182) gene and identification of six novel mutations in early onset AD families. Nat Genet 11: 219-222. doi: 10.1038/ng1095-219
|
| [4] |
Hardy J, Selkoe DJ (2002) The amyloid hypothesis of Alzheimer's disease: progress and problems on the road to therapeutics. Science 297: 353-356. doi: 10.1126/science.1072994
|
| [5] |
Weksler ME, Gouras G, Relkin NR, et al. (2005) The immune system, amyloid-beta peptide, and Alzheimer's disease. Immunol Rev 205: 244-256. doi: 10.1111/j.0105-2896.2005.00264.x
|
| [6] |
Hass MR, Sato C, Kopan R, et al. (2009) Presenilin: RIP and beyond. Semin Cell Dev Biol 20: 201-210. doi: 10.1016/j.semcdb.2008.11.014
|
| [7] |
de Calignon A, Fox LM, Pitstick R, et al. (2010) Caspase activation precedes and leads to tangles. Nature 464: 1201-1204. doi: 10.1038/nature08890
|
| [8] |
Sleegers K, Lambert JC, Bertram L, et al. (2010) The pursuit of susceptibility genes for Alzheimer's disease: progress and prospects. Trends Genet 26: 84-93. doi: 10.1016/j.tig.2009.12.004
|
| [9] |
Bertram L, Tanzi RE (2008) Thirty years of Alzheimer's disease genetics: the implications of systematic meta-analyses. Nat Rev Neurosci 9: 768-778. doi: 10.1038/nrn2494
|
| [10] |
Selkoe DJ, Hardy J (2016) The amyloid hypothesis of Alzheimer's disease at 25 years. EMBO Mol Med2016: e201606210. doi: 10.15252/emmm.201606210
|
| [11] |
Jones L, Holmans PA, Hamshere ML, et al. (2010) Genetic evidence implicates the immune system and cholesterol metabolism in the aetiology of Alzheimer's disease. PLoS One 5: e13950. doi: 10.1371/journal.pone.0013950
|
| [12] |
Porcellini E, Carbone I, Ianni M, et al. (2010) Alzheimer's disease gene signature says: beware of brain viral infections. Immun Ageing 7: 16. doi: 10.1186/1742-4933-7-16
|
| [13] |
Wozniak MA, Itzhaki RF, Shipley SJ, et al. (2007) Herpes simplex virus infection causes cellular beta-amyloid accumulation and secretase upregulation. Neurosci Lett 429: 95-100. doi: 10.1016/j.neulet.2007.09.077
|
| [14] |
Wozniak MA, Mee AP, Itzhaki RF (2009) Herpes simplex virus type 1 DNA is located within Alzheimer's disease amyloid plaques. J Pathol 217: 131-138. doi: 10.1002/path.2449
|
| [15] |
Santana S, Recuero M, Bullido MJ, et al. (2012) Herpes simplex virus type I induces the accumulation of intracellular beta-amyloid in autophagic compartments and the inhibition of the non-amyloidogenic pathway in human neuroblastoma cells. Neurobiol Aging 33: 430.e419-433. doi: 10.1016/j.neurobiolaging.2010.12.010
|
| [16] |
Seshadri S, Fitzpatrick AL, Ikram MA, et al. (2010) Genome-wide analysis of genetic loci associated with Alzheimer disease. JAMA 303: 1832-1840. doi: 10.1001/jama.2010.574
|
| [17] |
Carter CJ (2010) Alzheimer's Disease: A Pathogenetic Autoimmune Disorder Caused by Herpes Simplex in a Gene-Dependent Manner. Int J Alzheimers Dis 2010: 140539. doi: 10.4061/2010/140539
|
| [18] |
Tolia A, De Strooper B (2009) Structure and function of γ-secretase. Semin Cell Dev Biol 20: 211-218. doi: 10.1016/j.semcdb.2008.10.007
|
| [19] |
Edbauer D, Winkler E, Regula JT, et al. (2003) Reconstitution of gamma-secretase activity. Nat Cell Biol 5: 486-488. doi: 10.1038/ncb960
|
| [20] |
Kimberly WT, LaVoie MJ, Ostaszewski BL, et al. (2003) Gamma-secretase is a membrane protein complex comprised of presenilin, nicastrin, Aph-1, and Pen-2. Proc Natl Acad Sci U S A 100: 6382-6387. doi: 10.1073/pnas.1037392100
|
| [21] |
Takasugi N, Tomita T, Hayashi I, et al. (2003) The role of presenilin cofactors in the gamma-secretase complex. Nature 422: 438-441. doi: 10.1038/nature01506
|
| [22] |
Fraering PC, Ye W, Strub JM, et al. (2004) Purification and characterization of the human gamma-secretase complex. Biochemistry 43: 9774-9789. doi: 10.1021/bi0494976
|
| [23] |
Winkler E, Hobson S, Fukumori A, et al. (2009) Purification, pharmacological modulation, and biochemical characterization of interactors of endogenous human gamma-secretase. Biochemistry 48: 1183-1197. doi: 10.1021/bi801204g
|
| [24] |
Sato T, Diehl TS, Narayanan S, et al. (2007) Active gamma-secretase complexes contain only one of each component. J Biol Chem 282: 33985-33993. doi: 10.1074/jbc.M705248200
|
| [25] |
Hebert SS, Serneels L, Dejaegere T, et al. (2004) Coordinated and widespread expression of gamma-secretase in vivo: evidence for size and molecular heterogeneity. Neurobiol Dis 17: 260-272. doi: 10.1016/j.nbd.2004.08.002
|
| [26] |
Shirotani K, Edbauer D, Prokop S, et al. (2004) Identification of distinct gamma-secretase complexes with different APH-1 variants. J Biol Chem 279: 41340-41345. doi: 10.1074/jbc.M405768200
|
| [27] |
Beel AJ, Sanders CR (2008) Substrate specificity of gamma-secretase and other intramembrane proteases. Cell Mol Life Sci 65: 1311-1334. doi: 10.1007/s00018-008-7462-2
|
| [28] |
Coen K, Annaert W (2010) Presenilins: how much more than gamma-secretase?! Biochem Soc Trans 38: 1474-1478. doi: 10.1042/BST0381474
|
| [29] |
McCarthy JV, Twomey C, Wujek P (2009) Presenilin-dependent regulated intramembrane proteolysis and γ-secretase activity. Cell Mol Life Sci 66: 1534-1555. doi: 10.1007/s00018-009-8435-9
|
| [30] |
Wolfe MS (2009) Intramembrane-cleaving Proteases. J Biol Chem 284: 13969-13973. doi: 10.1074/jbc.R800039200
|
| [31] |
Brown MS, Ye J, Rawson RB, et al. (2000) Regulated intramembrane proteolysis: a control mechanism conserved from bacteria to humans. Cell 100: 391-398. doi: 10.1016/S0092-8674(00)80675-3
|
| [32] |
McCarthy JV, Twomey C, Wujek P (2009) Presenilin-dependent regulated intramembrane proteolysis and gamma-secretase activity. Cell Mol Life Sci 66: 1534-1555. doi: 10.1007/s00018-009-8435-9
|
| [33] |
Chhibber-Goel J, Coleman-Vaughan C, Agrawal V, et al. (2016) gamma-Secretase Activity Is Required for Regulated Intramembrane Proteolysis of Tumor Necrosis Factor (TNF) Receptor 1 and TNF-mediated Pro-apoptotic Signalling. J Biol Chem 291: 5971-5985. doi: 10.1074/jbc.M115.679076
|
| [34] | Elzinga BM, Twomey C, Powell JC, et al. (2009) Interleukin-1 receptor type 1 is a substrate for gamma-secretase-dependent regulated intramembrane proteolysis. J Biol Chem 284: 1394-1409. |
| [35] |
Kuhn PH, Marjaux E, Imhof A, et al. (2007) Regulated intramembrane proteolysis of the interleukin-1 receptor II by alpha-, beta-, and gamma-secretase. J Biol Chem 282: 11982-11995. doi: 10.1074/jbc.M700356200
|
| [36] |
Chalaris A, Gewiese J, Paliga K, et al. (2010) ADAM17-mediated shedding of the IL6R induces cleavage of the membrane stub by gamma-secretase. Biochim Biophys Acta 1803: 234-245. doi: 10.1016/j.bbamcr.2009.12.001
|
| [37] |
Schulte A, Schulz B, Andrzejewski MG, et al. (2007) Sequential processing of the transmembrane chemokines CX3CL1 and CXCL16 by alpha- and gamma-secretases. Biochem Biophys Res Commun 358: 233-240. doi: 10.1016/j.bbrc.2007.04.100
|
| [38] |
Donoviel DB, Hadjantonakis AK, Ikeda M, et al. (1999) Mice lacking both presenilin genes exhibit early embryonic patterning defects. Genes Dev 13: 2801-2810. doi: 10.1101/gad.13.21.2801
|
| [39] |
Xia X, Qian S, Soriano S, et al. (2001) Loss of presenilin 1 is associated with enhanced beta-catenin signalling and skin tumorigenesis. Proc Natl Acad Sci U S A 98: 10863-10868. doi: 10.1073/pnas.191284198
|
| [40] |
Guo Q, Fu W, Sopher BL, et al. (1999) Increased vulnerability of hippocampal neurons to excitotoxic necrosis in presenilin-1 mutant knock-in mice. Nat Med 5: 101-106. doi: 10.1038/4789
|
| [41] |
Feng R, Rampon C, Tang YP, et al. (2001) Deficient neurogenesis in forebrain-specific presenilin-1 knockout mice is associated with reduced clearance of hippocampal memory traces. Neuron 32: 911-926. doi: 10.1016/S0896-6273(01)00523-2
|
| [42] |
Wang R, Dineley KT, Sweatt JD, et al. (2004) Presenilin 1 familial Alzheimer's disease mutation leads to defective associative learning and impaired adult neurogenesis. Neuroscience 126: 305-312. doi: 10.1016/j.neuroscience.2004.03.048
|
| [43] |
Herreman A, Hartmann D, Annaert W, et al. (1999) Presenilin 2 deficiency causes a mild pulmonary phenotype and no changes in amyloid precursor protein processing but enhances the embryonic lethal phenotype of presenilin 1 deficiency. Proc Natl Acad Sci U S A 96: 11872-11877. doi: 10.1073/pnas.96.21.11872
|
| [44] |
Tournoy J, Bossuyt X, Snellinx A, et al. (2004) Partial loss of presenilins causes seborrheic keratosis and autoimmune disease in mice. Hum Mol Genet 13: 1321-1331. doi: 10.1093/hmg/ddh151
|
| [45] |
Maraver A, Tadokoro CE, Badura ML, et al. (2007) Effect of presenilins in the apoptosis of thymocytes and homeostasis of CD8+ T cells. Blood 110: 3218-3225. doi: 10.1182/blood-2007-01-070359
|
| [46] |
Laky K, Fowlkes BJ (2007) Presenilins regulate αβ T cell development by modulating TCR signalling. J Exp Med 204: 2115-2129. doi: 10.1084/jem.20070550
|
| [47] |
Yagi T, Giallourakis C, Mohanty S, et al. (2008) Defective signal transduction in B lymphocytes lacking presenilin proteins. Proc Natl Acad Sci U S A 105: 979-984. doi: 10.1073/pnas.0707755105
|
| [48] |
Beglopoulos V, Sun X, Saura CA, et al. (2004) Reduced beta-amyloid production and increased inflammatory responses in presenilin conditional knock-out mice. J Biol Chem 279: 46907-46914. doi: 10.1074/jbc.M409544200
|
| [49] |
Jayadev S, Case A, Eastman AJ, et al. (2010) Presenilin 2 Is the Predominant γ-Secretase in Microglia and Modulates Cytokine Release. PLoS ONE 5: e15743. doi: 10.1371/journal.pone.0015743
|
| [50] |
Glenn G, van der Geer P (2008) Toll-like receptors stimulate regulated intramembrane proteolysis of the CSF-1 receptor through Erk activation. FEBS Lett 582: 911-915. doi: 10.1016/j.febslet.2008.02.029
|
| [51] |
Garlind A, Brauner A, Hojeberg B, et al. (1999) Soluble interleukin-1 receptor type II levels are elevated in cerebrospinal fluid in Alzheimer's disease patients. Brain Res 826: 112-116. doi: 10.1016/S0006-8993(99)01092-6
|
| [52] |
Dinarello CA (2011) Interleukin-1 in the pathogenesis and treatment of inflammatory diseases. Blood 117: 3720-3732. doi: 10.1182/blood-2010-07-273417
|
| [53] |
Twomey C, Qian S, McCarthy JV (2009) TRAF6 promotes ubiquitination and regulated intramembrane proteolysis of IL-1R1. Biochem Biophys Res Commun 381: 418-423. doi: 10.1016/j.bbrc.2009.02.051
|
| [54] |
Powell JC, Twomey C, Jain R, et al. (2009) Association between Presenilin-1 and TRAF6 modulates regulated intramembrane proteolysis of the p75NTR neurotrophin receptor. J Neurochem 108: 216-230. doi: 10.1111/j.1471-4159.2008.05763.x
|
| [55] | Curtis BM, Widmer MB, deRoos P, et al. (1990) IL-1 and its receptor are translocated to the nucleus. J Immunol 144: 1295-1303. |
| [56] |
Levine SJ (2008) Molecular Mechanisms of Soluble Cytokine Receptor Generation. J Biol Chem 283: 14177-14181. doi: 10.1074/jbc.R700052200
|
| [57] |
Frykman S, Hur JY, Franberg J, et al. (2010) Synaptic and endosomal localization of active gamma-secretase in rat brain. PLoS One 5: e8948. doi: 10.1371/journal.pone.0008948
|
| [58] | Meckler X, Checler F (2016) Presenilin 1 and Presenilin 2 Target gamma-Secretase Complexes to Distinct Cellular Compartments. J Biol ChemM115: 708297. |
| [59] |
Doody RS, Raman R, Farlow M, et al. (2013) A phase 3 trial of semagacestat for treatment of Alzheimer's disease. N Engl J Med 369: 341-350. doi: 10.1056/NEJMoa1210951
|
| [60] |
Goodbourn S, Didcock L, Randall RE (2000) Interferons: cell signalling, immune modulation, antiviral response and virus countermeasures. J Gen Virol 81: 2341-2364. doi: 10.1099/0022-1317-81-10-2341
|
| [61] |
Saleh AZ, Fang AT, Arch AE, et al. (2004) Regulated proteolysis of the IFNaR2 subunit of the interferon-alpha receptor. Oncogene 23: 7076-7086. doi: 10.1038/sj.onc.1207955
|
| [62] |
Garton KJ, Gough PJ, Raines EW (2006) Emerging roles for ectodomain shedding in the regulation of inflammatory responses. J Leukoc Biol 79: 1105-1116. doi: 10.1189/jlb.0106038
|
| [63] |
Mambole A, Baruch D, Nusbaum P, et al. (2008) The cleavage of neutrophil leukosialin (CD43) by cathepsin G releases its extracellular domain and triggers its intramembrane proteolysis by presenilin/gamma-secretase. J Biol Chem 283: 23627-23635. doi: 10.1074/jbc.M710286200
|
| [64] |
Cui W, Ke JZ, Zhang Q, et al. (2006) The intracellular domain of CD44 promotes the fusion of macrophages. Blood 107: 796-805. doi: 10.1182/blood-2005-05-1902
|
| [65] |
Carey BW, Kim DY, Kovacs DM (2007) Presenilin/gamma-secretase and alpha-secretase-like peptidases cleave human MHC Class I proteins. Biochem J 401: 121-127. doi: 10.1042/BJ20060847
|
| [66] |
Bonifati DM, Kishore U (2007) Role of complement in neurodegeneration and neuroinflammation. Mol Immunol 44: 999-1010. doi: 10.1016/j.molimm.2006.03.007
|
| [67] |
Veerhuis R, Nielsen HM, Tenner AJ (2011) Complement in the brain. Mol Immunol 48: 1592-1603. doi: 10.1016/j.molimm.2011.04.003
|
| [68] |
Fonseca MI, Chu SH, Berci AM, et al. (2011) Contribution of complement activation pathways to neuropathology differs among mouse models of Alzheimer's disease. J Neuroinflammation 8: 4. doi: 10.1186/1742-2094-8-4
|
| [69] |
Webster SD, Galvan MD, Ferran E, et al. (2001) Antibody-Mediated Phagocytosis of the Amyloid β-Peptide in Microglia Is Differentially Modulated by C1q. J Immunol 166: 7496-7503. doi: 10.4049/jimmunol.166.12.7496
|
| [70] |
Fonseca MI, Zhou J, Botto M, et al. (2004) Absence of C1q leads to less neuropathology in transgenic mouse models of Alzheimer's disease. J Neurosci 24: 6457-6465. doi: 10.1523/JNEUROSCI.0901-04.2004
|
| [71] |
Fraser DA, Pisalyaput K, Tenner AJ (2010) C1q enhances microglial clearance of apoptotic neurons and neuronal blebs, and modulates subsequent inflammatory cytokine production. J Neurochem 112: 733-743. doi: 10.1111/j.1471-4159.2009.06494.x
|
| [72] |
Rogers J, Li R, Mastroeni D, et al. (2006) Peripheral clearance of amyloid beta peptide by complement C3-dependent adherence to erythrocytes. Neurobiol Aging 27: 1733-1739. doi: 10.1016/j.neurobiolaging.2005.09.043
|
| [73] |
Akiyama H, Barger S, Barnum S, et al. (2000) Inflammation and Alzheimer's disease. Neurobiol Aging 21: 383-421. doi: 10.1016/S0197-4580(00)00124-X
|
| [74] |
Spitzer P, Herrmann M, Klafki H-W, et al. (2010) Phagocytosis and LPS alter the maturation state of β-amyloid precursor protein and induce different Aβ peptide release signatures in human mononuclear phagocytes. J Neuroinflammation 7: 59-59. doi: 10.1186/1742-2094-7-59
|
| [75] |
Morgan D, Gordon MN, Tan J, et al. (2005) Dynamic complexity of the microglial activation response in transgenic models of amyloid deposition: implications for Alzheimer therapeutics. J Neuropathol Exp Neurol 64: 743-753. doi: 10.1097/01.jnen.0000178444.33972.e0
|
| [76] |
Streit WJ (2004) Microglia and Alzheimer's disease pathogenesis. J Neurosci Res 77: 1-8. doi: 10.1002/jnr.20093
|
| [77] | Trudler D, Farfara D, Frenkel D (2010) Toll-Like Receptors Expression and Signalling in Glia Cells in Neuro-Amyloidogenic Diseases: Towards Future Therapeutic Application. Mediators of Inflammation 2010: 12. |
| [78] |
Kumar H, Kawai T, Akira S (2011) Pathogen recognition by the innate immune system. Int Rev Immunol 30: 16-34. doi: 10.3109/08830185.2010.529976
|
| [79] |
Carty M, Bowie AG (2011) Evaluating the role of Toll-like receptors in diseases of the central nervous system. Biochem Pharmacol 81: 825-837. doi: 10.1016/j.bcp.2011.01.003
|
| [80] |
Chen F, Hasegawa H, Schmitt-Ulms G, et al. (2006) TMP21 is a presenilin complex component that modulates [gamma]-secretase but not [epsiv]-secretase activity. Nature 440: 1208-1212. doi: 10.1038/nature04667
|
| [81] | Iribarren P, Chen K, Hu J, et al. (2005) CpG-containing oligodeoxynucleotide promotes microglial cell uptake of amyloid beta 1-42 peptide by up-regulating the expression of the G-protein- coupled receptor mFPR2. FASEBJ 19: 2032-2034. |
| [82] |
Jin JJ, Kim HD, Maxwell JA, et al. (2008) Toll-like receptor 4-dependent upregulation of cytokines in a transgenic mouse model of Alzheimer's disease. J Neuroinflammation 5: 23. doi: 10.1186/1742-2094-5-23
|
| [83] |
Herber DL, Mercer M, Roth LM, et al. (2007) Microglial activation is required for Abeta clearance after intracranial injection of lipopolysaccharide in APP transgenic mice. J Neuroimmune Pharmacol 2: 222-231. doi: 10.1007/s11481-007-9069-z
|
| [84] |
DiCarlo G, Wilcock D, Henderson D, et al. (2001) Intrahippocampal LPS injections reduce Abeta load in APP+PS1 transgenic mice. Neurobiol Aging 22: 1007-1012. doi: 10.1016/S0197-4580(01)00292-5
|
| [85] |
Richard KL, Filali M, Prefontaine P, et al. (2008) Toll-like receptor 2 acts as a natural innate immune receptor to clear amyloid beta 1-42 and delay the cognitive decline in a mouse model of Alzheimer's disease. J Neurosci 28: 5784-5793. doi: 10.1523/JNEUROSCI.1146-08.2008
|
| [86] |
Jana M, Palencia CA, Pahan K (2008) Fibrillar amyloid-beta peptides activate microglia via TLR2: implications for Alzheimer's disease. J Immunol 181: 7254-7262. doi: 10.4049/jimmunol.181.10.7254
|
| [87] |
Frank S, Copanaki E, Burbach GJ, et al. (2009) Differential regulation of toll-like receptor mRNAs in amyloid plaque-associated brain tissue of aged APP23 transgenic mice. Neurosci Lett 453: 41-44. doi: 10.1016/j.neulet.2009.01.075
|
| [88] |
Michaud J-P, Richard KL, Rivest S (2011) MyD88-adaptor protein acts as a preventive mechanism for memory deficits in a mouse model of Alzheimer's disease. Mol Neurodegener 6: 5-5. doi: 10.1186/1750-1326-6-5
|
| [89] |
Cui JG, Li YY, Zhao Y, et al. (2010) Differential regulation of interleukin-1 receptor-associated kinase-1 (IRAK-1) and IRAK-2 by microRNA-146a and NF-kappaB in stressed human astroglial cells and in Alzheimer disease. J Biol Chem 285: 38951-38960. doi: 10.1074/jbc.M110.178848
|
| [90] |
Standridge JB (2006) Vicious cycles within the neuropathophysiologic mechanisms of Alzheimer's disease. Curr Alzheimer Res 3: 95-108. doi: 10.2174/156720506776383068
|
| [91] |
Lue LF, Walker DG, Rogers J (2001) Modeling microglial activation in Alzheimer's disease with human postmortem microglial cultures. Neurobiol Aging 22: 945-956. doi: 10.1016/S0197-4580(01)00311-6
|
| [92] | Yan Q, Zhang J, Liu H, et al. (2003) Anti-inflammatory drug therapy alters beta-amyloid processing and deposition in an animal model of Alzheimer's disease. J Neurosci 23: 7504-7509. |
| [93] |
Yamamoto M, Kiyota T, Horiba M, et al. (2007) Interferon-γ and Tumor Necrosis Factor-α Regulate Amyloid-β Plaque Deposition and β-Secretase Expression in Swedish Mutant APP Transgenic Mice. Am J Pathol 170: 680-692. doi: 10.2353/ajpath.2007.060378
|
| [94] |
Kong Q, Peterson TS, Baker O, et al. (2009) Interleukin-1beta enhances nucleotide-induced and alpha-secretase-dependent amyloid precursor protein processing in rat primary cortical neurons via up-regulation of the P2Y(2) receptor. J Neurochem 109: 1300-1310. doi: 10.1111/j.1471-4159.2009.06048.x
|
| [95] |
Sheng JG, Zhu SG, Jones RA, et al. (2000) Interleukin-1 Promotes Expression and Phosphorylation of Neurofilament and tau Proteins in Vivo. Exp Neurol 163: 388-391. doi: 10.1006/exnr.2000.7393
|
| [96] |
Moses GS, Jensen MD, Lue LF, et al. (2006) Secretory PLA2-IIA: a new inflammatory factor for Alzheimer's disease. J Neuroinflammation 3: 28. doi: 10.1186/1742-2094-3-28
|
| [97] |
Rezai-Zadeh K, Gate D, Gowing G, et al. (2011) How to Get from Here to There: Macrophage Recruitment in Alzheimer's Disease. Curr Alzheimer Res 8: 156-163. doi: 10.2174/156720511795256017
|
| [98] |
Shaftel SS, Kyrkanides S, Olschowka JA, et al. (2007) Sustained hippocampal IL-1 beta overexpression mediates chronic neuroinflammation and ameliorates Alzheimer plaque pathology. J Clin Invest 117: 1595-1604. doi: 10.1172/JCI31450
|
| [99] |
Rogers J, Strohmeyer R, Kovelowski CJ, et al. (2002) Microglia and inflammatory mechanisms in the clearance of amyloid beta peptide. Glia 40: 260-269. doi: 10.1002/glia.10153
|
| [100] |
Jutras I, Laplante A, Boulais J, et al. (2005) Gamma-secretase is a functional component of phagosomes. J Biol Chem 280: 36310-36317. doi: 10.1074/jbc.M504069200
|
| [101] |
Kiyota T, Yamamoto M, Xiong H, et al. (2009) CCL2 Accelerates Microglia-Mediated Aβ Oligomer Formation and Progression of Neurocognitive Dysfunction. PLoS ONE 4: e6197. doi: 10.1371/journal.pone.0006197
|
| [102] |
El Khoury J, Toft M, Hickman SE, et al. (2007) Ccr2 deficiency impairs microglial accumulation and accelerates progression of Alzheimer-like disease. Nat Med 13: 432-438. doi: 10.1038/nm1555
|
| [103] |
De Strooper B, Chavez Gutierrez L (2015) Learning by failing: ideas and concepts to tackle gamma-secretases in Alzheimer's disease and beyond. Annu Rev Pharmacol Toxicol 55: 419-437. doi: 10.1146/annurev-pharmtox-010814-124309
|
| [104] | Beher D, Clarke EE, Wrigley JD, et al. (2004) Selected non-steroidal anti-inflammatory drugs and their derivatives target gamma-secretase at a novel site. Evidence for an allosteric mechanism. J Biol Chem 279: 43419-43426. |
| [105] |
Kukar T, Golde TE (2008) Possible mechanisms of action of NSAIDs and related compounds that modulate gamma-secretase cleavage. Curr Top Med Chem 8: 47-53. doi: 10.2174/156802608783334042
|
| [106] |
Wong GT, Manfra D, Poulet FM, et al. (2004) Chronic treatment with the gamma-secretase inhibitor LY-411,575 inhibits beta-amyloid peptide production and alters lymphopoiesis and intestinal cell differentiation. J Biol Chem 279: 12876-12882. doi: 10.1074/jbc.M311652200
|
| [107] |
Jack C, Berezovska O, Wolfe MS, et al. (2001) Effect of PS1 deficiency and an APP gamma-secretase inhibitor on Notch1 signalling in primary mammalian neurons. Brain Res Mol Brain Res 87: 166-174. doi: 10.1016/S0169-328X(01)00010-9
|
| [108] |
Geling A, Steiner H, Willem M, et al. (2002) A gamma-secretase inhibitor blocks Notch signalling in vivo and causes a severe neurogenic phenotype in zebrafish. EMBO Rep 3: 688-694. doi: 10.1093/embo-reports/kvf124
|
| [109] | Micchelli CA, Esler WP, Kimberly WT, et al. (2003) Gamma-secretase/presenilin inhibitors for Alzheimer's disease phenocopy Notch mutations in Drosophila. FASEB J 17: 79-81. |
| [110] |
Henley DB, Sundell KL, Sethuraman G, et al. (2014) Safety profile of semagacestat, a gamma-secretase inhibitor: IDENTITY trial findings. Curr Med Res Opin 30: 2021-2032. doi: 10.1185/03007995.2014.939167
|
| [111] |
Panza F, Frisardi V, Imbimbo BP, et al. (2010) REVIEW: gamma-Secretase inhibitors for the treatment of Alzheimer's disease: The current state. CNS Neurosci Ther 16: 272-284. doi: 10.1111/j.1755-5949.2010.00164.x
|
| [112] |
Frisardi V, Solfrizzi V, Imbimbo PB, et al. (2010) Towards disease-modifying treatment of Alzheimer's disease: drugs targeting beta-amyloid. Curr Alzheimer Res 7: 40-55. doi: 10.2174/156720510790274400
|
| [113] |
Bergmans BA, De Strooper B (2010) gamma-secretases: from cell biology to therapeutic strategies. Lancet Neurol 9: 215-226. doi: 10.1016/S1474-4422(09)70332-1
|
| [114] | Samson K (2010) NerveCenter: Phase III Alzheimer trial halted: Search for therapeutic biomarkers continues. Ann Neurol 68: A9-a12. |
| [115] |
Gillman KW, Starrett JE, Jr., Parker MF, et al. (2010) Discovery and Evaluation of BMS-708163, a Potent, Selective and Orally Bioavailable gamma-Secretase Inhibitor. ACS Med Chem Lett 1: 120-124. doi: 10.1021/ml1000239
|
| [116] |
De Strooper B (2014) Lessons from a failed gamma-secretase Alzheimer trial. Cell 159: 721-726. doi: 10.1016/j.cell.2014.10.016
|
| [117] |
Cummings J (2010) What Can Be Inferred from the Interruption of the Semagacestat Trial for Treatment of Alzheimer's Disease? Biol Psychiatry 68: 876-878. doi: 10.1016/j.biopsych.2010.09.020
|
| [118] |
Qyang Y, Chambers SM, Wang P, et al. (2004) Myeloproliferative disease in mice with reduced presenilin gene dosage: effect of gamma-secretase blockage. Biochemistry 43: 5352-5359. doi: 10.1021/bi049826u
|
| [119] |
Imbimbo BP (2008) Therapeutic potential of gamma-secretase inhibitors and modulators. Curr Top Med Chem 8: 54-61. doi: 10.2174/156802608783334015
|
| [120] |
Pul R, Dodel R, Stangel M (2011) Antibody-based therapy in Alzheimer's disease. Expert Opin Biol Ther 11: 343-357. doi: 10.1517/14712598.2011.552884
|
| [121] |
Sarazin M, Dorothee G, de Souza LC, et al. (2013) Immunotherapy in Alzheimer's disease: do we have all the pieces of the puzzle? Biol Psychiatry 74: 329-332. doi: 10.1016/j.biopsych.2013.04.011
|
| [122] |
Brodaty H, Breteler MM, Dekosky ST, et al. (2011) The world of dementia beyond 2020. J Am Geriatr Soc 59: 923-927. doi: 10.1111/j.1532-5415.2011.03365.x
|
| [123] |
Yamada K, Yabuki C, Seubert P, et al. (2009) Abeta immunotherapy: intracerebral sequestration of Abeta by an anti-Abeta monoclonal antibody 266 with high affinity to soluble Abeta. J Neurosci 29: 11393-11398. doi: 10.1523/JNEUROSCI.2021-09.2009
|
| [124] |
Schenk D, Barbour R, Dunn W, et al. (1999) Immunization with amyloid-[beta] attenuates Alzheimer-disease-like pathology in the PDAPP mouse. Nature 400: 173-177. doi: 10.1038/22124
|
| [125] |
Lemere CA, Spooner ET, Leverone JF, et al. (2003) Amyloid-beta immunization in Alzheimer's disease transgenic mouse models and wildtype mice. Neurochem Res 28: 1017-1027. doi: 10.1023/A:1023203122036
|
| [126] |
Fu HJ, Liu B, Frost JL, et al. (2010) Amyloid-beta immunotherapy for Alzheimer's disease. CNS Neurol Disord Drug Targets 9: 197-206. doi: 10.2174/187152710791012017
|
| [127] | Robert R, Dolezal O, Waddington L, et al. (2009) Engineered antibody intervention strategies for Alzheimer's disease and related dementias by targeting amyloid and toxic oligomers. Protein Eng Des Sel 22: 199-208. |
| [128] |
Marin-Argany M, Rivera-Hernandez G, Marti J, et al. (2011) An anti-Abeta (amyloid beta) single-chain variable fragment prevents amyloid fibril formation and cytotoxicity by withdrawing Abeta oligomers from the amyloid pathway. Biochem J 437: 25-34. doi: 10.1042/BJ20101712
|
| [129] |
Panza F, Frisardi V, Imbimbo BP, et al. (2011) Anti-beta-amyloid immunotherapy for Alzheimer's disease: focus on bapineuzumab. Curr Alzheimer Res 8: 808-817. doi: 10.2174/156720511798192718
|
| [130] |
Lambracht-Washington D, Qu BX, Fu M, et al. (2011) DNA immunization against amyloid beta 42 has high potential as safe therapy for Alzheimer's disease as it diminishes antigen-specific Th1 and Th17 cell proliferation. Cell Mol Neurobiol 31: 867-874. doi: 10.1007/s10571-011-9680-7
|
| [131] |
Salloway S, Sperling R, Brashear HR (2014) Phase 3 trials of solanezumab and bapineuzumab for Alzheimer's disease. N Engl J Med 370: 1460. doi: 10.1056/NEJMc1402193
|
| [132] |
Salloway S, Sperling R, Fox NC, et al. (2014) Two phase 3 trials of bapineuzumab in mild-to-moderate Alzheimer's disease. N Engl J Med 370: 322-333. doi: 10.1056/NEJMoa1304839
|
| [133] |
Krishnamurthy PK, Sigurdsson EM (2011) Therapeutic applications of antibodies in non-infectious neurodegenerative diseases. New Biotechnol 28: 511-517. doi: 10.1016/j.nbt.2011.03.020
|
| [134] |
Britschgi M, Wyss-Coray T (2009) Blood protein signature for the early diagnosis of Alzheimer disease. Arch Neurol 66: 161-165. doi: 10.1001/archneurol.2008.530
|
| [135] |
Lindberg C, Chromek M, Ahrengart L, et al. (2005) Soluble interleukin-1 receptor type II, IL-18 and caspase-1 in mild cognitive impairment and severe Alzheimer's disease. Neurochem Int 46: 551-557. doi: 10.1016/j.neuint.2005.01.004
|
| [136] |
Reddy MM, Wilson R, Wilson J, et al. (2011) Identification of Candidate IgG Biomarkers for Alzheimer's Disease via Combinatorial Library Screening. Cell 144: 132-142. doi: 10.1016/j.cell.2010.11.054
|
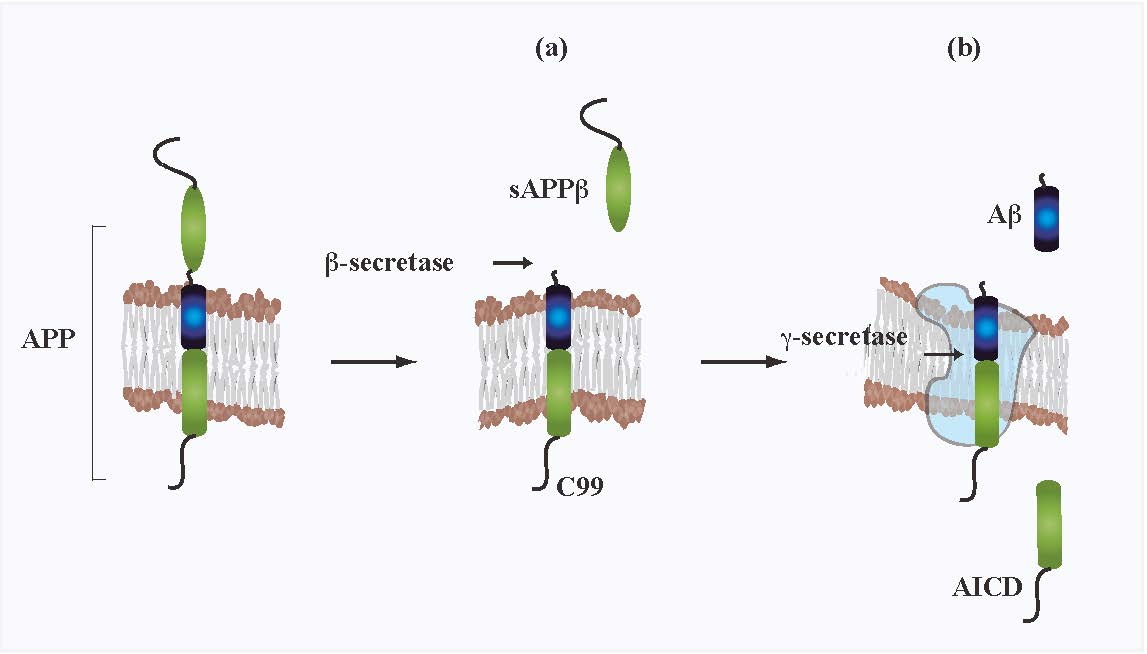
Figures(1)

Tara P. Hurst, Caroline Coleman-Vaughan, Indu Patwal, Justin V. McCarthy. Regulated intramembrane proteolysis, innate immunity and therapeutic targets in Alzheimer’s disease[J]. AIMS Molecular Science, 2016, 3(2): 138-157. doi: 10.3934/molsci.2016.2.138







 DownLoad:
DownLoad: