Citation: Sandip Bhattacharya, Debaprasad Das, Hafizur Rahaman. Analysis of temperature dependent power supply voltage drop in graphene nanoribbon and Cu based power interconnects[J]. AIMS Materials Science, 2016, 3(4): 1493-1506. doi: 10.3934/matersci.2016.4.1493

| [1] | Yamato Y, Yoneda T, Hatayama K, et al. (2012) A fast and accurate per-cell dynamic IR-drop estimation method for at-speed scan test pattern validation. IEEE International Test Conference (ITC). |
| [2] | Nithin SK, Shanmugam G, Chandrasekar S (2010) Dynamic voltage (IR) drop analysis and design closure: Issues and challenges. 11th International Symposium on Quality Electronic Design (ISQED). |
| [3] | Kumar A, Anis M (2010) IR-Drop Aware Clustering Technique for Robust Power Grid in FPGAs. IEEE T VLSI Syst 19: 1181–1191. |
| [4] | Vijayakumar A, Patil VC, Paladugu G, et al. (2014) On pattern generation for maximizing IR drop. 15th International Symposium on Quality Electronic Design (ISQED). |
| [5] | International Technology Roadmap for Semiconductors (ITRS-2013) Reports, 2013. Available from: http://www.itrs2.net/reports.html. |
| [6] | Naeemi A, Meindl JD (2008) Performance Benchmarking for Graphene Nanoribbon, Carbon Nanotube, and Cu Interconnects. International Interconnect Technology Conference (IITC). |
| [7] |
Naeemi A, Meindl JD (2009) Compact Physics-Based Circuit Models for Graphene Nano-ribbon Interconnects. IEEE T Electron Dev 56: 1822–1833. doi: 10.1109/TED.2009.2026122

|
| [8] |
Naeemi A, Meindl JD (2007) Conductance Modeling for Graphene Nanoribbon (GNR) Interconnects. IEEE Electr Device L 28: 428–431. doi: 10.1109/LED.2007.895452
|
| [9] |
Xu C, Li H, Banerjee K (2009) Modeling, Analysis, and Design of Graphene Nano-Ribbon Interconnects. IEEE T Electron Dev 56: 1567–1578. doi: 10.1109/TED.2009.2024254
|
| [10] |
Nasiri SH, Moravvej-Farshi MK, Faez R (2010) Stability Analysis in Graphene Nanoribbon Interconnects. IEEE Electr Device L 31: 1458–1460. doi: 10.1109/LED.2010.2079312
|
| [11] | Tanachutiwat S, Liu SH, Geer R, Wei W (2009) Monolithic grapheme nanoribbon electronics for interconnect performance improvement. IEEE International Symposium on Circuits and Systems. |
| [12] | Das D, Rahaman H (2012) Modeling of IR-Drop induced delay fault in CNT and GNR power distribution networks. 5th International Conference on Computers and Devices for Communication (CODEC). |
| [13] | Das D, Rahaman H (2015) Carbon Nanotube and Graphene Nanoribbon Interconnects, 1st Eds., New York, CRC Press, 37–78. |
| [14] | Das D, Rahaman H (2012) Simultaneous switching noise and IR drop in graphene nanoribbon power distribution networks. 12th IEEE Conference on Nanotechnology (IEEE-NANO). |
| [15] | Alizadeh A, Sarvari R (2015) Temperature-Dependent Comparison Between Delay of CNT and Copper Interconnects. IEEE T VLSI Syst 9: 1–1. |
| [16] |
Fratini S, Guinea F (2008) Substrate-limited electron dynamics in graphene. Phys Rev B Condens Matter Mater Phys 77: 195415. doi: 10.1103/PhysRevB.77.195415
|
| [17] |
Fuchs K (1938) Conduction electrons in thin metallic films. In Proc Cambridge Phil Soc 34: 100. doi: 10.1017/S0305004100019952
|
| [18] |
Sondheimer EH (1952) The mean free path of electrons in metals. Adv Phys 1: 1–42. doi: 10.1080/00018735200101151
|
| [19] |
Mayadas AF, Shatzkes M (1970) Electrical resistivity model for polycrystalline films: the case of arbitrary reflection at external surfaces. Phys Rev B Condens Matter Mater Phys 1: 1382–1389. doi: 10.1103/PhysRevB.1.1382
|
| [20] |
Goetsch RJ, Anand VK, Pandey A, et al. (2012) Structural, thermal, magnetic, and electronic transport properties of the LaNi2(Ge1−xPx)2 system. Phys Rev B Condens Matter Mater Phys 85: 054517. doi: 10.1103/PhysRevB.85.054517
|
| [21] | Blatt FJ (1968) Physics of Electronic Conduction in Solids. McGraw-Hill, New York. |
| [22] | Bid A, Bora A, Raychaudhuri AK (2006) Temperature dependence of the resistance of metallic nanowires of diameter ≥15 nm: applicability of Bloch-Grüneisen theorem. Phys Rev B Condens Matter Mater Phys 3: 1–9. |
| [23] | Gusakova D, Ryzhanova N, Vedyayev A, et al. (2004) Influence of s-d scattering on the electron density of states in ferromagnet/superconductor bilayer. J Magn Magn Mater 42: 873–882. |
| [24] | Predictive Technology Model, 2008. Available from: http://ptm.asu.edu. |
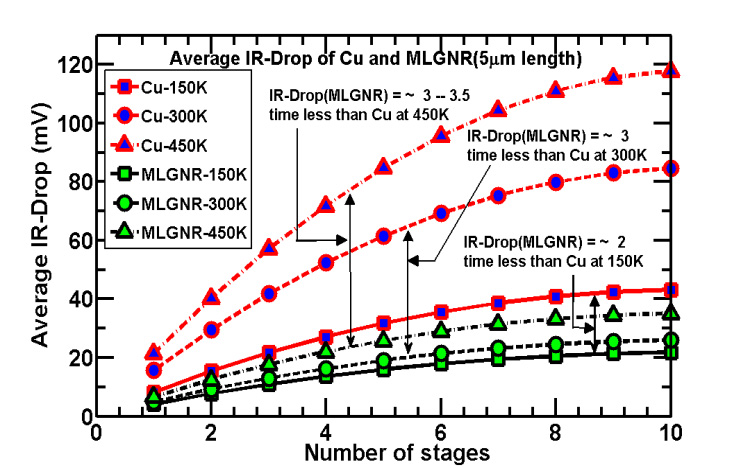
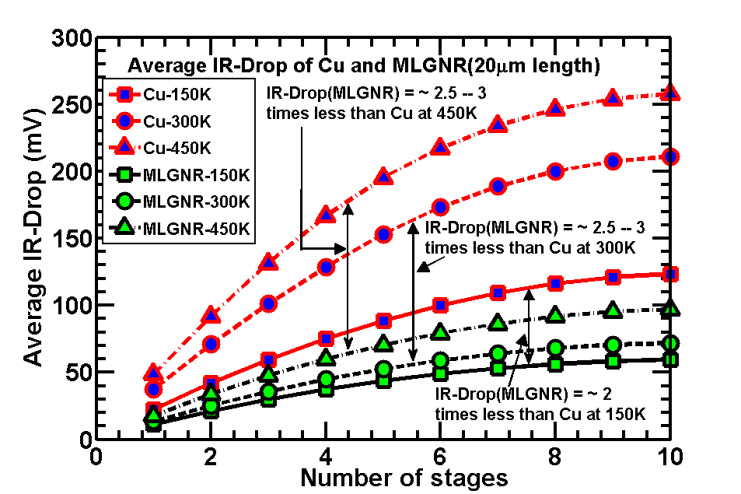
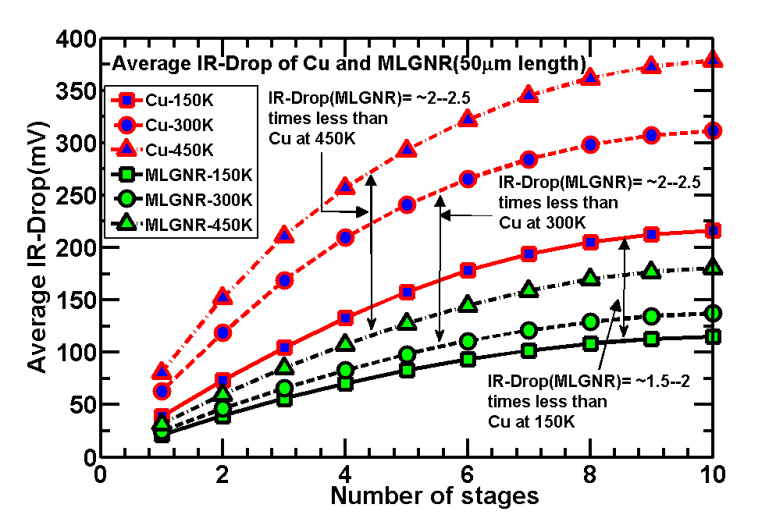
Figures(7) / Tables(5)

Sandip Bhattacharya, Debaprasad Das, Hafizur Rahaman. Analysis of temperature dependent power supply voltage drop in graphene nanoribbon and Cu based power interconnects[J]. AIMS Materials Science, 2016, 3(4): 1493-1506. doi: 10.3934/matersci.2016.4.1493







 DownLoad:
DownLoad: