Alzheimer's disease stands as one of the most widespread neurodegenerative conditions associated with aging, giving rise to dementia and posing significant public health challenges. Mathematical models are considered as valuable tools to gain insights into the mechanisms underlying the onset, progression, and potential therapeutic approaches for AD. In this paper, we introduce a mathematical model for AD that employs the fractal fractional operator in the Caputo sense to characterize the temporal dynamics of key cell populations. This model encompasses essential elements, including amyloid-β ($\mathbb{ A_\beta }$), neurons, astroglia and microglia. Using the fractal fractional operator, we have established the existence and uniqueness of solutions for the model under consideration, employing Leray-Schaefer's theorem and the Banach fixed-point methods. Utilizing functional techniques, we have analyzed the proposed model stability under the Ulam-Hyers condition. The suggested model has been numerically simulated by using a fractional Adams-Bashforth approach, which involves a two-step Lagrange polynomial. For numerical simulations, different ranges of fractional order values and fractal dimensions are considered. This new fractal fractional operator in the form of the Caputo derivative was determined to yield better results than an ordinary integer order. Various outcomes are shown graphically by for different fractal dimensions and arbitrary orders.
Citation: Pooja Yadav, Shah Jahan, Kottakkaran Sooppy Nisar. Analysis of fractal-fractional Alzheimer's disease mathematical model in sense of Caputo derivative[J]. AIMS Public Health, 2024, 11(2): 399-419. doi: 10.3934/publichealth.2024020
Alzheimer's disease stands as one of the most widespread neurodegenerative conditions associated with aging, giving rise to dementia and posing significant public health challenges. Mathematical models are considered as valuable tools to gain insights into the mechanisms underlying the onset, progression, and potential therapeutic approaches for AD. In this paper, we introduce a mathematical model for AD that employs the fractal fractional operator in the Caputo sense to characterize the temporal dynamics of key cell populations. This model encompasses essential elements, including amyloid-β ($\mathbb{ A_\beta }$), neurons, astroglia and microglia. Using the fractal fractional operator, we have established the existence and uniqueness of solutions for the model under consideration, employing Leray-Schaefer's theorem and the Banach fixed-point methods. Utilizing functional techniques, we have analyzed the proposed model stability under the Ulam-Hyers condition. The suggested model has been numerically simulated by using a fractional Adams-Bashforth approach, which involves a two-step Lagrange polynomial. For numerical simulations, different ranges of fractional order values and fractal dimensions are considered. This new fractal fractional operator in the form of the Caputo derivative was determined to yield better results than an ordinary integer order. Various outcomes are shown graphically by for different fractal dimensions and arbitrary orders.

| [1] | Patterson C (2018) The state of the art of dementia research: New frontiers. World Alzheimer Report . |
| [2] |
Fotuhi M, Hachinski V, Whitehouse PJ (2009) Changing perspectives regarding late-life dementia. Nat Rev Neurol 5: 649-658. https://doi.org/10.1038/nrneurol.2009.175 
|
| [3] |
Nebel RA, Aggarwal NT, Barnes LL, et al. (2018) Understanding the impact of sex and gender in Alzheimer's disease: a call to action. Alzheimers Dement 14: 1171-1183. https://doi.org/10.1016/j.jalz.2018.04.008
|
| [4] |
Zárate S, Stevnsner T, Gredilla R (2017) Role of estrogen and other sex hormones in brain aging. Neuroprotection and DNA repair. Front Aging Neurosci 9: 430. https://doi.org/10.3389/fnagi.2017.00430
|
| [5] | What Are the Symptoms of High Estrogen? (2022). Available from: https://www.medicalnewstoday.com/articles/323280#treatment |
| [6] |
Navakkode S, Gaunt JR, Pavon MV, et al. (2021) Sex-specific accelerated decay in time/activity-dependent plasticity and associative memory in an animal model of Alzheimer's disease. Aging Cell 20: e13502. https://doi.org/10.1111/acel.13502
|
| [7] |
Jung YJ, Chung WS (2018) Phagocytic roles of glial cells in healthy and diseased brains. Biomol Ther 26: 350. https://doi.org/10.4062/biomolther.2017.133
|
| [8] |
Selkoe DJ (1997) Alzheimer's Disease–Genotypes, Phenotype, and Treatments. Sci 275: 630-631. https://doi.org/10.1126/science.275.5300.630
|
| [9] |
Holtzman DM, Bales KR, Tenkova T, et al. (2000) Apolipoprotein E isoform-dependent amyloid deposition and neuritic degeneration in a mouse model of Alzheimer's disease. Pro Natl Acad Sci 97: 2892-2897. https://doi.org/10.1073/pnas.050004797
|
| [10] |
Perry VH, Nicoll JA, Holmes C (2010) Microglia in neurodegenerative disease. Nat Rev Neurol 6: 193-201. https://doi.org/10.1038/nrneurol.2010.17
|
| [11] |
Lue LF, Kuo YM, Beach T, et al. (2010) Microglia activation and anti-inflammatory regulation in Alzheimer's disease. Mol Neurobiol 41: 115-128. https://doi.org/10.1007/s12035-010-8106-8
|
| [12] |
Neumann J, Sauerzweig S, Rönicke R, et al. (2008) Microglia cells protect neurons by direct engulfment of invading neutrophil granulocytes: a new mechanism of CNS immune privilege. J Neurosci 28: 5965-5975. https://doi.org/10.1523/JNEUROSCI.0060-08.2008
|
| [13] |
Pang Y, Campbell L, Zheng B, et al. (2010) Lipopolysaccharide-activated microglia induce death of oligodendrocyte progenitor cells and impede their development. Neurosci 166: 464-475. https://doi.org/10.1016/j.neuroscience.2009.12.040
|
| [14] | Granas A, Dugundji J (2003) Fixed point theory. New York: Springer 14. https://doi.org/10.1007/978-0-387-21593-8 |
| [15] |
Varnum MM, Ikezu T (2012) The classification of microglial activation phenotypes on neurodegeneration and regeneration in Alzheimer's disease brain. Arch Immunol Ther Exp 60: 251-266. https://doi.org/10.1007/s00005-012-0181-2
|
| [16] |
Proctor CJ, Boche D, Gray DA, et al. (2013) Investigating interventions in alzheimer's disease with computer simulation models. PLoS One 8: e73631. https://doi.org/10.1371/journal.pone.0073631
|
| [17] |
Hadjichrysanthou C, Ower AK, de Wolf F, et al. (2018) Alzheimer's Disease Neuroimaging Initiative. The development of a stochastic mathematical model of Alzheimer's disease to help improve the design of clinical trials of potential treatments. PLoS One 13: e0190615. https://doi.org/10.1371/journal.pone.0190615
|
| [18] |
Perez C, Ziburkus J, Ullah G (2016) Analyzing and modeling the dysfunction of inhibitory neurons in Alzheimer's disease. PLoS One 11: e0168800. https://doi.org/10.1371/journal.pone.0168800
|
| [19] |
Macdonald A, Pritchard D (2000) A mathematical model of Alzheimer's disease and the ApoE gene. ASTIN Bull 30: 69-110. https://doi.org/10.2143/AST.30.1.504627
|
| [20] |
Hane F, Augusta C, Bai O (2018) A hierarchical Bayesian model to predict APOE4 genotype and the age of Alzheimer's disease onset. PLoS One 13: e0200263. https://doi.org/10.1371/journal.pone.0200263
|
| [21] |
Jack CR, Holtzman DM (2013) Biomarker modeling of Alzheimer's disease. Neuron 80: 1347-1358. http://dx.doi.org/10.1016/j.neuron.2013.12.003
|
| [22] |
Young AL, Oxtoby NP, Daga P, et al. (2014) A data-driven model of biomarker changes in sporadic Alzheimer's disease. Brain 137: 2564-2577. https://doi.org/10.1093/brain/awu176
|
| [23] |
Puri IK, Li L (2010) Mathematical modeling for the pathogenesis of Alzheimer's disease. PLoS One 5: e15176. https://doi.org/10.1371/journal.pone.0015176
|
| [24] |
Hao W, Friedman A (2016) Mathematical model on Alzheimer's disease. BMC Syst Biol 10: 1-8. https://doi.org/10.1186/s12918-016-0348-2
|
| [25] | Bertsch M, Franchi B, Marcello N, et al. (2017) Alzheimer's disease: a mathematical model for onset and progression. Math Med Biol 34: 193-214. https://doi.org/10.1093/imammb/dqw003 |
| [26] |
Vosoughi A, Sadigh-Eteghad S, Ghorbani M, et al. (2020) Mathematical models to shed light on amyloid-beta and tau protein dependent pathologies in Alzheimer's disease. Neurosci 424: 45-57. https://doi.org/10.1016/j.neuroscience.2019.09.017
|
| [27] |
Helal M, Hingant E, Pujo-Menjouet L, et al. (2014) Alzheimer's disease: analysis of a mathematical model incorporating the role of prions. J Math Biol 69: 1207-1235. https://doi.org/10.1007/s00285-013-0732-0
|
| [28] | Caputo M, Fabrizio M (2015) A new definition of fractional derivative without singular kernel. Prog Fract Differ Appl 1: 73-85. http://dx.doi.org/10.12785/pfda/010201 |
| [29] | Losada J, Nieto JJ (2015) Properties of a new fractional derivative without singular kernel. Progr Fract Differ Appl 1: 87-92. http://dx.doi.org/10.12785/pfda/010202 |
| [30] |
Yadav P, Jahan S, Shah K, et al. (2023) Fractional-order modelling and analysis of diabetes mellitus: Utilizing the Atangana-Baleanu Caputo (ABC) operator. Alex Eng J 81: 200-209. https://doi.org/10.1016/j.aej.2023.09.006
|
| [31] |
Atangana A, Khan MA (2019) Validity of fractal derivative to capturing chaotic attractors. Chaos Solit 126: 50-59. https://doi.org/10.1016/j.chaos.2019.06.002
|
| [32] |
He JH (2018) Fractal calculus and its geometrical explanation. Results Phys 10: 272-276. https://doi.org/10.1016/j.rinp.2018.06.011
|
| [33] |
Gomez-Aguilar JF, Cordova-Fraga T, Abdeljawad T, et al. (2020) Analysis of fractal–fractional malaria transmission model. Fractals 28: 2040041. https://doi.org/10.1142/S0218348X20400411
|
| [34] |
Ali Z, Rabiei F, Shah K, et al. (2021) Qualitative analysis of fractal-fractional order COVID-19 mathematical model with case study of Wuhan. Alex Eng J 60: 477-489. https://doi.org/10.1016/j.aej.2020.09.020
|
| [35] |
Qu H, Ur Rahman M, Arfan M, et al. (2022) Investigating fractal-fractional mathematical model of Tuberculosis (TB) under fractal-fractional Caputo operator. Fractals 30: 2240126. https://doi.org/10.1142/S0218348X22401260
|
| [36] |
Rivers-Auty J, Mather AE, Peters R, et al. (2020) Anti-inflammatories in Alzheimer's disease-potential therapy or spurious correlate?. Brain Commun 2: fcaa109. https://doi.org/10.1093/braincomms/fcaa109
|
| [37] | Lynch AM, Murphy KJ, Deighan BF, et al. (2010) The impact of glial activation in the aging brain. Aging Dis 1: 262. |
| [38] |
Babu NR, Balasubramaniam P (2023) Master-slave synchronization for glucose–insulin metabolism of type-1 diabetic Mellitus model based on new fractal–fractional order derivative. Math Comput Simul 204: 282-301. https://doi.org/10.1016/j.matcom.2022.08.014
|
| [39] | Babu NR, Balasubramaniam P, Ratnavelu K (2023) Existence and uniqueness for a new perturbed chaotic jerk circuit model based on fractal-fractional derivative. AIP Conf 2756. |
| [40] | Shah K, Abdalla B, Abdeljawad T, et al. (2024) A Fractal-Fractional Order Model to Study Multiple Sclerosis: A Chronic Disease. Fractals 2440010. https://doi.org/10.1142/S0218348X24400103 |
| [41] | Shah K, Abdeljawad T (2024) Study of radioactive decay process of uranium atoms via fractals-fractional analysis. S Afr J Chem Eng 48: 63-70. https://doi.org/10.1016/j.sajce.2024.01.003 |
| [42] |
Khan ZA, Shah K, Abdalla B, et al. (2023) A numerical study of complex dynamics of a chemostat model under fractal-fractional derivative. Fractals 31: 2340181. https://doi.org/10.1142/S0218348X23401813
|
| [43] |
Shah K, Abdeljawad T (2023) On complex fractal-fractional order mathematical modeling of CO 2 emanations from energy sector. Phys Scr 99: 015226. https://doi.org/10.1088/1402-4896/ad1286
|













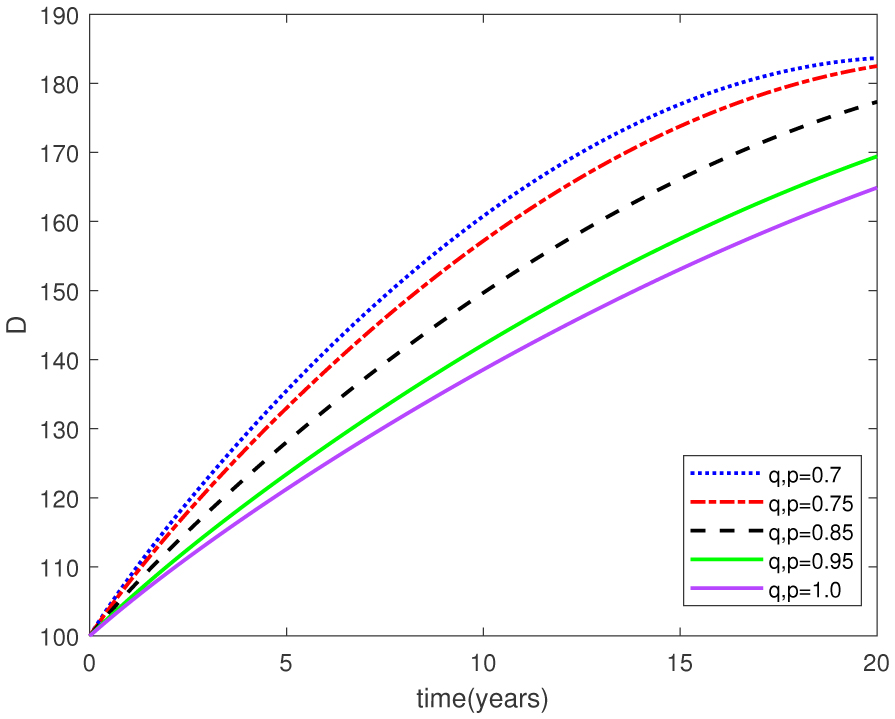
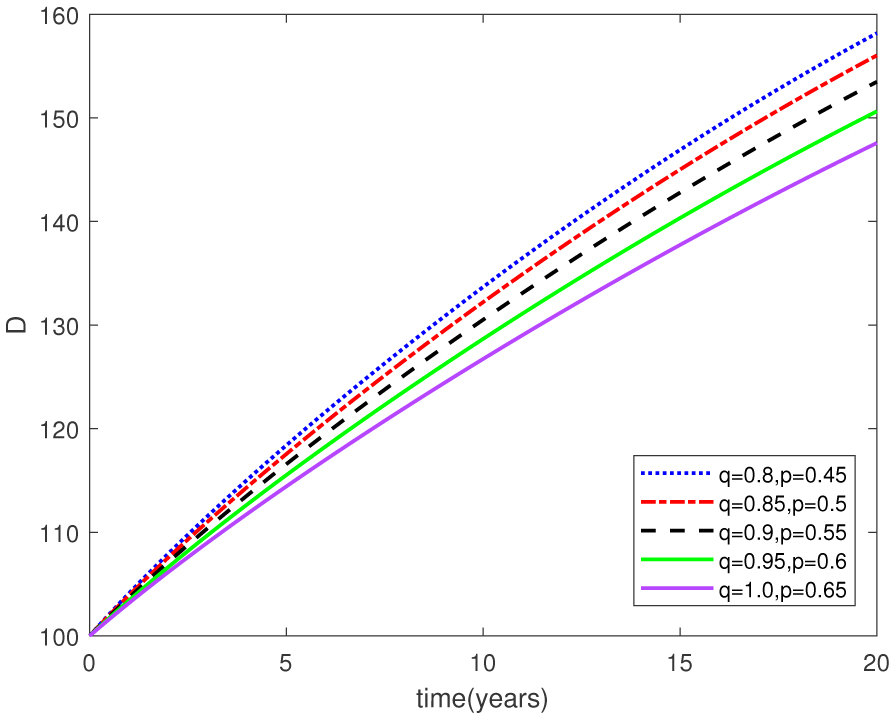
Figures(15) / Tables(1)

Pooja Yadav, Shah Jahan, Kottakkaran Sooppy Nisar. Analysis of fractal-fractional Alzheimer's disease mathematical model in sense of Caputo derivative[J]. AIMS Public Health, 2024, 11(2): 399-419. doi: 10.3934/publichealth.2024020







 DownLoad:
DownLoad: