Citation: Alireza Sayyidmousavi, Katrin Rohlf. Stochastic simulations of the Schnakenberg model with spatial inhomogeneities using reactive multiparticle collision dynamics[J]. AIMS Mathematics, 2019, 4(6): 1805-1823. doi: 10.3934/math.2019.6.1805

| [1] |
D. T. Gillespie, Exact stochastic simulation of coupled chemical reactions, J. Phys. Chem., 81 (1977), 2340-2361. doi: 10.1021/j100540a008

|
| [2] |
D. T. Gillespie, Approximate accelerated stochastic simulation of reacting systems, J. Phys. Chem., 115 (2001), 1716-1733. doi: 10.1063/1.1378322
|
| [3] |
A. Stundzia, C. Lumsden, Stochastic simulation of coupled reaction-diffusion processes, J. Comput. Phys., 127 (1996), 196-207. doi: 10.1006/jcph.1996.0168
|
| [4] |
S. Isaacson, C. Peskin, Incorporating diffusion in complex geometries into stochastic chemical kinetics simulations, SIAM J. Sci. Comput., 28 (2006), 47-74. doi: 10.1137/040605060
|
| [5] | D. Fange, J. Elf, Noise-induced min phenotypes in E. coli, PLoS Comput. Biol., 2 (2006), e80. |
| [6] |
L. Ferm, A. Hellander, P. Lotstedt, An adaptive algorithm for simulation of stochastic reaction-diffusion processes, J. Comput. Phys., 229 (2010), 343-360. doi: 10.1016/j.jcp.2009.09.030
|
| [7] | J. M. A. Padgett, S. Ilie, An adaptive tau-leaping method for stochastic simulations of reaction-diffusion systems, AIP Adv., 6 (2016), 035217. |
| [8] | R. Strehl, S. Ilie, An adaptive tau-leaping method for stochastic systems with slow and fast dynamics, J. Chem. Phys., 143 (2015), 234108. |
| [9] | C. A. Smith, C. A. Yates, Spatially extended hybrid methods: A review, J. R. Soc. Interface, 15 (2018), 20170931. |
| [10] |
M. B. Flegg, S. J. Chapman, R. Erban, The two regime method for optimizing stochastic reaction-diffusion simulations, J. R. Soc. Interface, 9 (2012), 859-868. doi: 10.1098/rsif.2011.0574
|
| [11] |
A. Hellander, S. Hellander, P. Lotstedt, Coupled mesoscopic and microscopic simulation of stochastic reaction-diffusion processes in mixed dimensions, Multiscale Model. Sim., 10 (2012), 585-611. doi: 10.1137/110832148
|
| [12] | M. B. Flegg, S. Hellander, R. Erban, Convergence of methods for coupling of microscopic and mesoscopic reaction-diffusion simulations, J. Chem. Phys., 289 (2015), 1-17. |
| [13] |
J. Hattne, D. Fange, J. Elf, Stochastic reaction-diffusion simulation with MesoRD, Bioinformatics, 21 (2005), 2923-2924. doi: 10.1093/bioinformatics/bti431
|
| [14] |
S. S. Andrews, D. Bray, Stochastic simulation of chemical reactions with spatial resolution and single molecule detail, Phys. Biol., 1 (2004), 137-151. doi: 10.1088/1478-3967/1/3/001
|
| [15] | J. S. van Zon, P. R. ten Wolde, Greens-function reaction dynamics: A particle-based approach for simulating biochemical networks in time and space, J. Chem. Phys., 123 (2005), 234910. |
| [16] | J. S. van Zon, P. R. ten Wolde, Simulating biochemical networks at the particle level and in time and space: Green's function reaction dynamics, Phys. Rev. Lett., 94 (2005), 128103. |
| [17] |
S. J. Chapman, R. Erban, S. A. Isaacson, Reactive boundary conditions as limits of interaction potentials for Brownian and Langevin dynamics, SIAM J. Appl. Math., 76 (2016), 368-390. doi: 10.1137/15M1030662
|
| [18] |
H. G. Othmer, S. R. Dunbar, W. Alt, Models of dispersal in biological systems, J. Math. Biol., 26 (1988), 263-298. doi: 10.1007/BF00277392
|
| [19] |
Y. Cao, R. Erban, Stochastic Turing patterns: Analysis of compartment-based approaches, B. Math. Biol., 76 (2014), 3051-3069. doi: 10.1007/s11538-014-0044-6
|
| [20] |
M. B. Flegg, Smoluchowski reaction kinetics for reactions of any order, SIAM J. Appl. Math., 76 (2016), 1403-1432. doi: 10.1137/15M1030509
|
| [21] |
A. Malevanets, R. Kapral, Mesoscopic model for solvent dynamics, J. Chem. Phys., 110 (1999), 8605-8613. doi: 10.1063/1.478857
|
| [22] |
A. Malevanets, R. Kapral, Solute molecular dynamics in a mesoscale solvent, J. Chem. Phys., 112 (2000), 7260-7269. doi: 10.1063/1.481289
|
| [23] |
K. Tucci, R. Kapral, Mesoscopic model for diffusion influenced reaction dynamics, J. Chem. Phys., 120 (2004), 8262-8270. doi: 10.1063/1.1690244
|
| [24] |
K. Tucci, R. Kapral, Mesoscopic multiparticle collision dynamics of reaction-diffusion fronts, J. Chem. Phys. B, 109 (2005), 21300-21304. doi: 10.1021/jp052701u
|
| [25] |
K. Rohlf, S. Fraser, R. Kapral, Reactive multiparticle collision dynamics, Comput. Phys. Commun., 179 (2008), 132-139. doi: 10.1016/j.cpc.2008.01.027
|
| [26] |
K. Rohlf, Stochastic phase-space description for reactions that change particle numbers, J. Math. Chem., 45 (2009), 141-160. doi: 10.1007/s10910-008-9373-8
|
| [27] | J. M. Yeomans, Mesoscale simulations: Lattice Boltzmann and particle algorithms, Physica A, 369 (2006), 159. |
| [28] | P. J. Hoogerbrugge, J. M. V. A. Koelman, Simulating microscopic hydrodynamic phenomena with dissipative particle dynamics, EPL, 19 (1992), 155. |
| [29] |
S. Bedkihal, J. C. Kumaradas, K. Rohlf, Steady flow through a constricted cylinder by multiparticle collision dynamics, Biomech. Model. Mechan., 12 (2013), 929-939. doi: 10.1007/s10237-012-0454-z
|
| [30] |
T. Akhter, K. Rohlf, Quantifying compressibility and slip in multiparticle collision (MPC) flow through a local constriction, Entropy, 16 (2014), 418-442. doi: 10.3390/e16010418
|
| [31] | K. Rohlf, Compressible slip flow through constricted cylinders with density-dependent viscosity, Dynam. Cont. Dis. Ser. B, 25 (2018), 233-257. |
| [32] | T. Ihle, D. M. Kroll, Stochastic rotation dynamics: A Galilean-invariant mesoscopic model for fluid flow, Phys. Rev. E, 63 (2001), 020201. |
| [33] | T. Ihle, D. M. Kroll, Stochastic rotation dynamics: I. Formalism, Galilean invariance, and Green-Kubo relations, Phys. Rev. E, 67 (2003), 066705. |
| [34] | T. Ihle, D. M. Kroll, Stochastic rotation dynamics: II. Transport coefficients, numerics, and long-time tails, Phys. Rev. E, 67 (2003), 066706. |
| [35] | H. Noguchi, G. Gompper, Transport coefficients of off-lattice mesoscale-hydrodynamics simulation techniques, Phys. Rev. E, 78 (2008), 016706. |
| [36] | E. Tüzel, T. Ihle, D. M. Kroll, Dynamic correlations in stochastic rotation dynamics, Phys. Rev. E, 74 (2006), 056702. |
| [37] | R. Strehl, K. Rohlf, Multiparticle collision dynamics for diffusion-influenced signaling pathways, Phys. Biol., 13 (2016), 046004. |
| [38] | A. Sayyidmousavi, K. Rohlf, Reactive multi-particle collision dynamics with reactive boundary conditions, Phys. Biol., 15 (2018), 046007. |
| [39] |
V. Mortazavi, M. Nosonovsky, Friction-induced pattern formation and Turing systems, Langmuir, 27 (2011), 4772-4779. doi: 10.1021/la200272x
|
| [40] |
D. A. Garzon-Alvarado, C. H. Galeano, J. M. Mantilla, Computational examples of reaction-convection-diffusion equations solution under the influence of fluid flow: A first example, Appl. Math. Model., 36 (2012), 5029-5045. doi: 10.1016/j.apm.2011.12.041
|
| [41] |
J. Wei, M. Winter, Flow-distributed spikes for Schnakenberg kinetics, J. Math. Biol., 64 (2012), 211-254. doi: 10.1007/s00285-011-0412-x
|
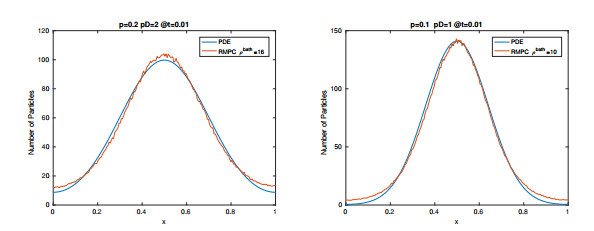
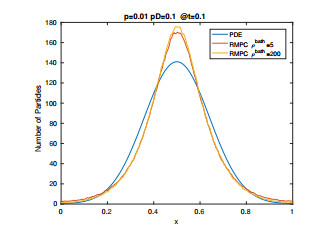
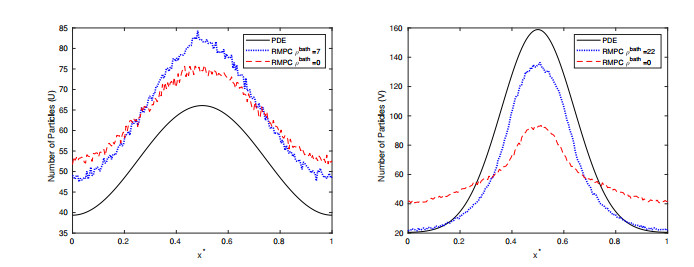
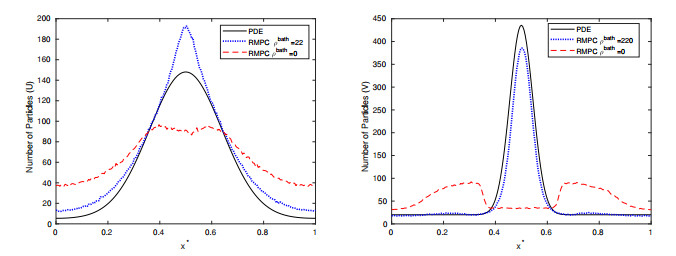
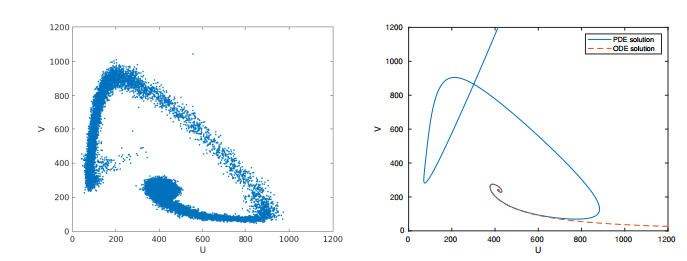
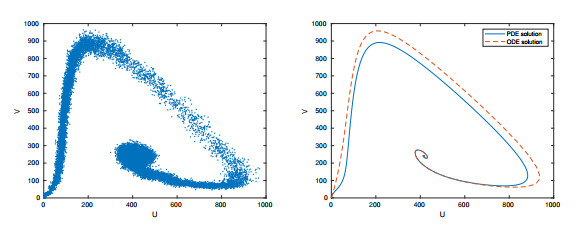
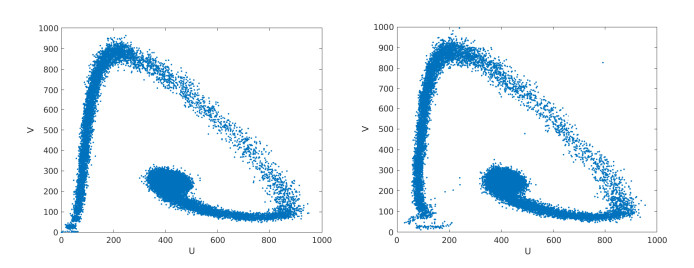
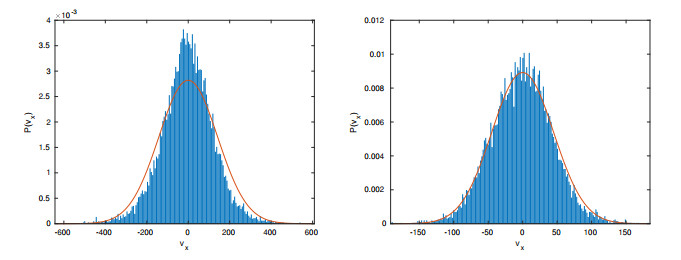
Figures(15) / Tables(2)

Alireza Sayyidmousavi, Katrin Rohlf. Stochastic simulations of the Schnakenberg model with spatial inhomogeneities using reactive multiparticle collision dynamics[J]. AIMS Mathematics, 2019, 4(6): 1805-1823. doi: 10.3934/math.2019.6.1805







 DownLoad:
DownLoad: