Citation: Judit Faus-Garriga, Isabel Novoa, Andrés Ozaita. mTOR signaling in proteostasis and its relevance to autism spectrum disorders[J]. AIMS Biophysics, 2017, 4(1): 63-89. doi: 10.3934/biophy.2017.1.63

| [1] | Gumeni S, Trougakos IP (2016) Cross talk of proteostasis and mitostasis in cellular homeodynamics, ageing, and disease. Oxidative Medicine and Cellular Longevity 2016: 4587691. |
| [2] | Ruegsegger C, Saxena S (2016) Proteostasis impairment in ALS. Brain Res S0006-8993: 30161–30165. |
| [3] | Pluquet O, Pourtier A, Abbadie C (2015) The unfolded protein response and cellular senescence. A review in the theme: cellular mechanisms of endoplasmic reticulum stress signaling in health and disease. Am J Physiol Cell Physiol 308: C415–C425. |
| [4] | Klein ME, Monday H, Jordan BA (2016) Proteostasis and RNA binding proteins in synaptic plasticity and in the pathogenesis of neuropsychiatric disorders. Neural Plast 2016: 3857934. |
| [5] |
Nakada C, Ritchie K, Oba Y, et al. (2003) Accumulation of anchored proteins forms membrane diffusion barriers during neuronal polarization. Nat Cell Biol 5: 626–632. doi: 10.1038/ncb1009

|
| [6] |
Guillery RW (2005) Observations of synaptic structures: origins of the neuron doctrine and its current status. Philos Trans R Soc Lond B Biol Sci 360: 1281–1307. doi: 10.1098/rstb.2003.1459
|
| [7] |
Citri A, Malenka RC (2008) Synaptic plasticity: multiple forms, functions, and mechanisms. Neuropsychopharmacology 33: 18–41. doi: 10.1038/sj.npp.1301559
|
| [8] |
Kessels HW, Malinow R (2009) Synaptic AMPA receptor plasticity and behavior. Neuron 61: 340–350. doi: 10.1016/j.neuron.2009.01.015
|
| [9] | Hardingham N, Dachtler J, Fox K (2013) The role of nitric oxide in pre-synaptic plasticity and homeostasis. Front Cell Neurosci 7: 190. |
| [10] |
Hashimotodani Y, Ohno-Shosaku T, Kano M (2007) Endocannabinoids and synaptic function in the CNS. Neuroscientist 13: 127–137. doi: 10.1177/1073858406296716
|
| [11] |
Casadio A, Martin KC, Giustetto M, et al. (1999) A transient, neuron-wide form of CREB-mediated long-term facilitation can be stabilized at specific synapses by local protein synthesis. Cell 99: 221–237. doi: 10.1016/S0092-8674(00)81653-0
|
| [12] | Huber KM,Kayser MS, Bear MF (2000) Role for rapid dendritic protein synthesis in hippocampal mGluR-dependent long-term depression. Science 288: 1254–1256. |
| [13] | Bradshaw KD, Emptage NJ, Bliss TV (2003) A role for dendritic protein synthesis in hippocampal late LTP. Eur J Neurosci 18: 3150–3152. |
| [14] |
Yin HH, Davis MI, Ronesi JA, et al. (2006) The role of protein synthesis in striatal long-term depression. J Neurosci 26: 11811–11820. doi: 10.1523/JNEUROSCI.3196-06.2006
|
| [15] |
Zhang Y, Nicholatos J, Dreier JR, et al. (2014) Coordinated regulation of protein synthesis and degradation by mTORC1. Nature 513: 440–443. doi: 10.1038/nature13492
|
| [16] |
Louros SR, Osterweil EK (2016) Perturbed proteostasis in autism spectrum disorders. J Neurochem 139: 1081–1092. doi: 10.1111/jnc.13723
|
| [17] | American Psychiatric Association, (2013) Diagnostic and Statistical Manual of Mental Disorders: DSM-5, Arlington, Virginia: USA American Psychiatric Association. |
| [18] | de la Torre-Ubieta L, Won H, Stein JL, et al. (2016) Advancing the understanding of autism disease mechanisms through genetics. Nat Med 22: 345–361. |
| [19] | Geschwind DH, State MW (2015) Gene hunting in autism spectrum disorder: on the path to precision medicine. Lancet Neurol 14: 1109–1120. |
| [20] |
Jaworski J, Sheng M (2006) The growing role of mTOR in neuronal development and plasticity. Mol Neurobiol 34: 205–219. doi: 10.1385/MN:34:3:205
|
| [21] |
Bockaert J, Marin P (2015) mTOR in brain physiology and pathologies. Physiol Rev 95: 1157–1187. doi: 10.1152/physrev.00038.2014
|
| [22] | Bramham CR, Wells DG (2007) Dendritic mRNA: transport, translation and function. Nat Rev Neurosci 8: 776–789. |
| [23] |
Wang DO, Kim SM, Zhao Y, et al. (2009) Synapse- and stimulus-specific local translation during long-term neuronal plasticity. Science 324: 1536–1540. doi: 10.1126/science.1173205
|
| [24] | Andreassi C, Riccio A (2009)To localize or not to localize: mRNA fate is in 3'UTRends. Trends Cell Biol 19: 465–474. |
| [25] | Besse F, Ephrussi A (2008) Translational control of localized mRNAs: restricting protein synthesis in space and time. Nat Rev Mol Cell Biol 9: 971–980. |
| [26] |
Redondo RL, Morris R (2011) Making memories last: the synaptic tagging and capture hypothesis. Nat Rev Neurosci 12: 17–30. doi: 10.1038/nrn2963
|
| [27] |
Richter JD, Klann E (2009) Making synaptic plasticity and memory last: mechanisms of translational regulation. Genes Dev 23: 1–11. doi: 10.1101/gad.1735809
|
| [28] |
Thomas MG, Pascual ML, Maschi D, et al. (2014) Synaptic control of local translation: the plot thickens with new characters. Cell Mol Life Sci 71: 2219–2239. doi: 10.1007/s00018-013-1506-y
|
| [29] |
Doyle M, Kiebler MA (2011) Mechanisms of dendritic mRNA transport and its role in synaptic tagging. EMBO J 30: 3540–3552. doi: 10.1038/emboj.2011.278
|
| [30] |
Mayford M, Baranes D, Podsypanina K, et al. (1996) The 3'-untranslated region of CaMKII alpha is a cis-acting signal for the localization and translation of mRNA in dendrites. Proc Natl Acad Sci USA 93: 13250–13255. doi: 10.1073/pnas.93.23.13250
|
| [31] |
Kislauskis EH, Li Z, Singer RH, et al. (1993) Isoform-specific 3'-untranslated sequences sort alpha-cardiac and beta-cytoplasmic actin messenger RNAs to different cytoplasmic compartments. J Cell Biol 123: 165–172. doi: 10.1083/jcb.123.1.165
|
| [32] | Blichenberg A, Schwanke B, Rehbein M, et al. (1999) Identification of a cis-acting dendritic targeting element in MAP2 mRNAs. J Neurosci 19: 8818–8829. |
| [33] |
Kobayashi H, Yamamoto S, Maruo T, et al. (2005) Identification of a cis-acting element required for dendritic targeting of activity-regulated cytoskeleton-associated protein mRNA. Eur J Neurosci 22: 2977–2984. doi: 10.1111/j.1460-9568.2005.04508.x
|
| [34] |
An JJ, Gharami K, Liao GY, et al. (2008) Distinct role of long 3' UTR BDNF mRNA in spine morphology and synaptic plasticity in hippocampal neurons. Cell 134: 175–187. doi: 10.1016/j.cell.2008.05.045
|
| [35] |
Glanzer J, Miyashiro KY, Sul JY, et al. (2005) RNA splicing capability of live neuronal dendrites. Proc Natl Acad Sci USA 102: 16859–16864. doi: 10.1073/pnas.0503783102
|
| [36] |
Chawla G, Lin CH, Han A, et al. (2009) Sam68 regulates a set of alternatively spliced exons during neurogenesis. Mol Cell Biol 29: 201–213. doi: 10.1128/MCB.01349-08
|
| [37] |
Matter N, Herrlich P, König H (2002) Signal-dependent regulation of splicing via phosphorylation of Sam68. Nature 420: 691–695. doi: 10.1038/nature01153
|
| [38] | Khaladkar M, Buckley PT, Lee MT, et al. (2013) Subcellular RNA sequencing reveals broad presence of cytoplasmic intron-sequence retaining transcripts in mouse and rat neurons. PLoS One 8: 1–13. |
| [39] | Buckley PT, Lee MT, Sul JY, et al. (2011) Cytoplasmic intron sequence-retaining transcripts can be dendritically targeted via ID element retrotransposons. Neuron 69: 877–884. |
| [40] | Buchan JR (2014) mRNP granules. Assembly, function, and connections with disease. RNA Biol 11: 1019–1030. |
| [41] | Fritzsche R, Karra D, Bennett KL, et al. (2013) Interactome of two diverse RNA granules links mRNA localization to translational repression in neurons. Cell Rep 5: 1749–1762. |
| [42] |
Ivanov PA, Chudinova EM, Nadezhdina ES (2003) Disruption of microtubules inhibits cytoplasmic ribonucleoprotein stress granule formation. Exp Cell Res 290: 227–233. doi: 10.1016/S0014-4827(03)00290-8
|
| [43] |
Hirokawa N (2006) mRNA transport in dendrites: RNA granules, motors, and tracks. J Neurosci 26: 7139–7142. doi: 10.1523/JNEUROSCI.1821-06.2006
|
| [44] |
Martin KC, Ephrussi A (2009) mRNA localization: gene expression in the spatial dimension. Cell 136: 719–730. doi: 10.1016/j.cell.2009.01.044
|
| [45] | Kapeli K, Yeo GW (2012) Genome-wide approaches to dissect the roles of RNA binding proteins in translational control: implications for neurological diseases. Front Neurosci 6: 144. |
| [46] |
Laggerbauer B, Ostareck D, Keidel EM, et al. (2001) Evidence that fragile X mental retardation protein is a negative regulator of translation. Hum Mol Genet 10: 329–338. doi: 10.1093/hmg/10.4.329
|
| [47] | Hüttelmaier S, Zenklusen D, Lederer M, et al. (2005) Spatial regulation of β-actin translation by Src-dependent phosphorylation of ZBP1. Nature 438: 512–515. |
| [48] | Darnell JC, Richter JD (2012) Cytoplasmic RNA-binding proteins and the control of complex brain function. Cold Spring Harb Perspect Biol 4: a012344. |
| [49] |
Klein ME, Younts TJ, Castillo PE, et al. (2013) RNA-binding protein Sam68 controls synapse number and local β-actin mRNA metabolism in dendrites. Proc Natl Acad Sci USA 110: 3125–3130. doi: 10.1073/pnas.1209811110
|
| [50] | Grange J, Belly A, Dupas S, et al. (2009) Specific interaction between Sam68 and neuronal mRNAs: implication for the activity-dependent biosynthesis of elongation factor eEF1A. J Neurosci Res 87: 12–25. |
| [51] | Eom T, Antar LN, Singer RH, et al. (2003) Localization of a beta-actin messenger ribonucleoprotein complex with zipcode-binding protein modulates the density of dendritic filopodia and filopodial synapses. J Neurosci 23: 10433–10444. |
| [52] |
Itoh M, Haga I, Li QH, et al. (2002) Identification of cellular mRNA targets for RNA-binding protein Sam68. Nucleic Acids Res 30: 5452–5464. doi: 10.1093/nar/gkf673
|
| [53] |
Jung MY, Lorenz L, Richter JD (2006) Translational control by neuroguidin, a eukaryotic initiation factor 4E and CPEB binding protein. Mol Cell Biol 26: 4277–4287. doi: 10.1128/MCB.02470-05
|
| [54] |
Richter JD, Sonenberg N (2005) Regulation of cap-dependent translation by eIF4E inhibitory proteins. Nature 433: 477–480. doi: 10.1038/nature03205
|
| [55] |
Ivshina M, Lasko P, Richter JD (2014) Cytoplasmic polyadenylation element binding proteins in development, health, and disease. Annu Rev Cell Dev Biol 30: 393–415. doi: 10.1146/annurev-cellbio-101011-155831
|
| [56] |
Huang YS, Jung MY, Sarkissian M, et al. (2002) N-methyl-D-aspartate receptor signaling results in Aurora kinase-catalyzed CPEB phosphorylation and alpha CaMKII mRNA polyadenylation at synapses. EMBO J 21: 2139–2148. doi: 10.1093/emboj/21.9.2139
|
| [57] |
Wang CF, Huang YS (2012) Calpain 2 activated through N-methyl-D-aspartic acid receptor signaling cleaves CPEB3 and abrogates CPEB3-repressed translation in neurons. Mol Cell Biol 32: 3321–3332. doi: 10.1128/MCB.00296-12
|
| [58] |
Siomi H, Siomi MC, Nussbaum RL, et al. (1993) The protein product of the fragile X gene, FMR1, has characteristics of an RNA-binding protein. Cell 74: 291–298. doi: 10.1016/0092-8674(93)90420-U
|
| [59] |
Verkerk AJ, Pieretti M, Sutcliffe JS, et al. (1991) Identification of a gene (FMR-1) containing a CGG repeat coincident with a breakpoint cluster region exhibiting length variation in fragile X syndrome. Cell 65: 905–914. doi: 10.1016/0092-8674(91)90397-H
|
| [60] | Tsuboi D, Kuroda K, Tanaka M, et al. (2015) Disrupted-in-schizophrenia 1 regulates transport of ITPR1 mRNA for synaptic plasticity. Nat Neurosc 18: 698–707. |
| [61] | Cohen TJ, Lee VM, Trojanowski JQ (2011) TDP-43 functions and pathogenic mechanisms implicated in TDP-43 proteinopathies. Trends Mol Med 17: 659–667. |
| [62] |
Koyama A, Sugai A, Kato T, et al. (2016) Increased cytoplasmic TARDBP mRNA in affected spinal motor neurons in ALS caused by abnormal autoregulation of TDP-43. Nucleic Acids Res 44: 5820–5836. doi: 10.1093/nar/gkw499
|
| [63] |
Udagawa T, Farny NG, Jakovcevski M, et al. (2013) Genetic and acute CPEB1 depletion ameliorate fragile X pathophysiology. Nat Med 19: 1473–1477. doi: 10.1038/nm.3353
|
| [64] |
Iacoangeli A, Tiedge H (2013) Translational control at the synapse: role of RNA regulators. Trends Biochem Sci 38: 47–55. doi: 10.1016/j.tibs.2012.11.001
|
| [65] |
Santini E, Huynh TN, Klann E (2014) Mechanisms of translation control underlying long-lasting synaptic plasticity and the consolidation of long-term memory. Prog Mol Biol Transl Sci 122: 131–167. doi: 10.1016/B978-0-12-420170-5.00005-2
|
| [66] |
Sonenberg N, Hinnebusch AG (2009) Regulation of translation initiation in eukaryotes: mechanisms and biological targets. Cell 136: 731–745. doi: 10.1016/j.cell.2009.01.042
|
| [67] |
Thoreen CC (2013) Many roads from mTOR to eIF4F. Biochem Soc Trans 41: 913–916. doi: 10.1042/BST20130082
|
| [68] |
Pyronnet S (2000) Phosphorylation of the capbinding protein eIF4E by the MAPK-activated protein kinase Mnk1. Biochem Pharmacol 60: 1237–1243. doi: 10.1016/S0006-2952(00)00429-9
|
| [69] |
Panja D, Dagyte G, Bidinosti M, et al. (2009) Novel translational control in Arc-dependent long term potentiation consolidation in vivo. J Biol Chem 284: 31498–31511. doi: 10.1074/jbc.M109.056077
|
| [70] |
Tang SJ, Reis G, Kang H, et al. (2002) A rapamycin-sensitive signaling pathway contributes to long-term synaptic plasticity in the hippocampus. Proc Natl Acad Sci USA 99: 467–472. doi: 10.1073/pnas.012605299
|
| [71] |
Vilchez D, Saez I, Dillin A (2014) The role of protein clearance mechanisms in organismal ageing and age-related diseases. Nat Commun 5: 5659. doi: 10.1038/ncomms6659
|
| [72] |
Yi JJ, Ehlers MD (2005) Ubiquitin and protein turnover in synapse function. Neuron 47: 629–632. doi: 10.1016/j.neuron.2005.07.008
|
| [73] | Hamilton AM, Zito K (2013) Breaking it down: the ubiquitin proteasome system in neuronal morphogenesis. Neural Plast 2013: 196848. |
| [74] | Bingol B, Schuman EM (2005) Synaptic protein degradation by the ubiquitin proteasome system. Curr Opin Neurobiol 15: 536–541. |
| [75] |
Ravid T, Hochstrasser M (2008) Diversity of degradation signals in the ubiquitin-proteasome system. Nat Rev Mol Cell Biol 9: 679–690. doi: 10.1038/nrm2468
|
| [76] |
Hegde AN (2010) The ubiquitin-proteasome pathway and synaptic plasticity. Learn Mem 17: 314–327. doi: 10.1101/lm.1504010
|
| [77] |
Bingol B, Sheng M (2011) Deconstruction for reconstruction: the role of proteolysis in neural plasticity and disease. Neuron 69: 22–32. doi: 10.1016/j.neuron.2010.11.006
|
| [78] |
Bingol B, Schuman EM (2006) Activity-dependent dynamics and sequestration of proteasomes in dendritic spines. Nature 441: 1144–1148. doi: 10.1038/nature04769
|
| [79] | Hegde AN (2016) Proteolysis, synaptic plasticity and memory. Neurobiol Learn Mem S1074-7427: 30178–30172. |
| [80] |
Li Q, Korte M, Sajikumar S (2016) Ubiquitin-proteasome system inhibition promotes long-term depression and synaptic tagging/capture. Cereb Cortex 26: 2541–2548. doi: 10.1093/cercor/bhv084
|
| [81] | Ciechanover A, Kwon YT (2015) Degradation of misfolded proteins in neurodegenerative diseases: therapeutic targets and strategies. ExpMol Med 47: e147. |
| [82] |
Tarpey PS, Raymond FL, O'Meara S, et al. (2007) Mutations in CUL4B, which encodes a ubiquitin E3 ligase subunit, cause an X-linked mental retardation syndrome associated with aggressive outbursts, seizures, relative macrocephaly, central obesity, hypogonadism, pescavus, and tremor. Am J Hum Genet 80: 345–352. doi: 10.1086/511134
|
| [83] |
Dong C, Bach SV, Haynes KA, et al. (2014) Proteasome modulates positive and negative translational regulators in long-term synaptic plasticity. J Neurosci 34: 3171–3182. doi: 10.1523/JNEUROSCI.3291-13.2014
|
| [84] |
Johnson CW, Melia TJ, Yamamoto A (2012) Modulating macroautophagy: a neuronal perspective. Future Med Chem 4: 1715–1731. doi: 10.4155/fmc.12.112
|
| [85] | Komatsu M, Waguri S, Chiba T, et al. (2006) Loss of autophagy in the central nervous system causes neurodegeneration in mice. Nature 2006 441: 880–884. |
| [86] |
Mizushima N, Komatsu M (2011) Autophagy: renovation of cells and tissues. Cell 147: 728–741. doi: 10.1016/j.cell.2011.10.026
|
| [87] |
Hu Z, Yang B, Mo X, et al. (2015) Mechanism and regulation of autophagy and its role in neuronal diseases. Mol Neurobiol 52: 1190–1209. doi: 10.1007/s12035-014-8921-4
|
| [88] |
Tang G, Gudsnuk K, Kuo SH, et al. (2014) Loss of mTOR-dependent macroautophagy causes autistic-like synaptic pruning deficits. Neuron 83: 1131–1143. doi: 10.1016/j.neuron.2014.07.040
|
| [89] |
Penzes P, Cahill ME, Jones KA, et al. (2011) Dendritic spine pathology in neuropsychiatric disorders. Nat Neurosci 14: 285–293. doi: 10.1038/nn.2741
|
| [90] |
Riccomagno MM, Kolodkin AL (2015) Sculpting neural circuits by axon and dendrite pruning. Annu Rev Cell Dev Biol 31: 779–805. doi: 10.1146/annurev-cellbio-100913-013038
|
| [91] |
Jung CH, Ro SH, Cao J, et al. (2010) mTOR regulation of autophagy. FEBS Lett 584: 1287–1295. doi: 10.1016/j.febslet.2010.01.017
|
| [92] |
Lipton JO, Sahin M (2014) The neurology of mTOR. Neuron 84: 275–291. doi: 10.1016/j.neuron.2014.09.034
|
| [93] | Roohi A, Hojjat-Farsangi M (2016) Recent Advances in targeting mTORsignaling pathway using small molecule inhibitors. J Drug Target 15: 1–37. |
| [94] |
Jacinto E, Loewith R, Schmidt A, et al. (2004) Mammalian TOR complex 2 controls the actin cytoskeleton and is rapamycin insensitive. Nat Cell Biol 6: 1122–1128. doi: 10.1038/ncb1183
|
| [95] |
Gaubitz C, Prouteau M, Kusmider B, et al. (2016) TORC2 structure and function. Trends Biochem Sci 41: 532–545. doi: 10.1016/j.tibs.2016.04.001
|
| [96] |
Huang W, Zhu PJ, Zhang S, et al. (2013) mTORC2 controls actin polymerization required for consolidation of long-term memory. Nat Neurosci 16: 441–448. doi: 10.1038/nn.3351
|
| [97] |
Johnson JL, Huang W, Roman G, et al. (2015) TORC2: a novel target for treating age-associated memory impairment. Sci Rep 5: 15193. doi: 10.1038/srep15193
|
| [98] |
Lenz G, Avruch J (2005) Glutamatergic regulation of the p70S6 kinase in primary mouse neurons. J Biol Chem 280: 38121–38124. doi: 10.1074/jbc.C500363200
|
| [99] |
Takei N, Inamura N, Kawamura M, et al. (2004) Brain-derived neurotrophic factor induces mammalian target of rapamycin-dependent local activation of translation machinery and protein synthesis in neuronal dendrites. J Neurosci 24: 9760–9769. doi: 10.1523/JNEUROSCI.1427-04.2004
|
| [100] |
Lee CC, Huang CC, Wu MY, et al. (2005) Insulin stimulates postsynaptic density-95 protein translation via the phosphoinositide 3-kinase-Akt-mammalian target of rapamycin signaling pathway. J Biol Chem 280: 18543–18550. doi: 10.1074/jbc.M414112200
|
| [101] |
Costa-Mattioli M, Monteggia LM (2013) mTOR complexes in neurodevelopmental and neuropsychiatric disorders. Nat Neurosci 16: 1537–1543. doi: 10.1038/nn.3546
|
| [102] |
Sosanya NM, Cacheaux LP, Workman ER, et al. (2015) Mammalian target of rapamycin (mTOR) tagging promotes dendritic branch variability through the capture of Ca2+/calmodulin-dependent protein kinase II α (CaMKIIα) mRNAs by the RNA-binding protein HuD. J Biol Chem 290: 16357–16371. doi: 10.1074/jbc.M114.599399
|
| [103] |
Meyuhas O, Kahan T (2015) The race to decipher the top secrets of TOP mRNAs. Biochim Biophys Acta 1849: 801–811. doi: 10.1016/j.bbagrm.2014.08.015
|
| [104] | Ehninger D, Han S, Shilyansky C, et al. (2008) Reversal of learning deficits in a Tsc2+/– mouse model of tuberous sclerosis. Nat Med 14: 843–848. |
| [105] |
Kwon CH, Luikart BW, Powell CM, et al. (2006) Pten regulates neuronal arborization and social interaction in mice. Neuron 50: 377–388. doi: 10.1016/j.neuron.2006.03.023
|
| [106] |
Hoeffer CA, Sanchez E, Hagerman RJ, et al. (2012) Altered mTOR signaling and enhanced CYFIP2 expression levels in subjects with fragile X syndrome. Genes Brain Behav 11: 332–341. doi: 10.1111/j.1601-183X.2012.00768.x
|
| [107] | Sharma A, Hoeffer CA, Takayasu Y, et al. (2010) Deregulation of mTOR signaling in fragile X syndrome. J Neurosci 30: 694–702. |
| [108] |
Ricciardi S, Boggio EM, Grosso S, et al. (2011) Reduced AKT/mTOR signaling and protein synthesis deregulation in a Rett syndrome animal model. Hum Mol Genet 20: 1182–1196. doi: 10.1093/hmg/ddq563
|
| [109] |
Ramocki MB, Tavyev YJ, Peters SU (2010) The MECP2 duplication syndrome. Am J Med Genet A 152A: 1079–1088. doi: 10.1002/ajmg.a.33184
|
| [110] |
Sun J, Liu Y, Moreno S, et al. (2015) Imbalanced mechanistic target of rapamycin C1 and C2 activity in the cerebellum of Angelman syndrome mice impairs motor function. J Neurosci 35: 4706–4718. doi: 10.1523/JNEUROSCI.4276-14.2015
|
| [111] |
Stornetta RL, Zhu JJ (2011) Ras and Rap signaling in synaptic plasticity and mental disorders. Neuroscientist 17: 54–78. doi: 10.1177/1073858410365562
|
| [112] |
Phillips M, Pozzo-Miller L (2015) Dendritic spine dysgenesis in autism related disorders. Neurosci Lett 601: 30–40. doi: 10.1016/j.neulet.2015.01.011
|
| [113] | Huber KM, Klann E, Costa-Mattioli M, et al. (2015) Deregulation of mammalian target of rapamycin signaling in mouse models of autism. J Neurosci 35: 13836–13842. |
| [114] |
Kelleher RJ III, Bear MF (2008) The autistic neuron: troubled translation? Cell 135: 401–406. doi: 10.1016/j.cell.2008.10.017
|
| [115] |
Zhou J, Blundell J, Ogawa S, et al. (2009) Pharmacological inhibition of mTORC1 suppresses anatomical, cellular, and behavioral abnormalities in neural-specific Pten knock-out mice. J Neurosci 29: 1773–1783. doi: 10.1523/JNEUROSCI.5685-08.2009
|
| [116] |
Busquets-Garcia A, Gomis-González M, Guegan T, et al. (2013) Targeting the endocannabinoid system in the treatment of fragile X syndrome. Nat Med 19: 603–607. doi: 10.1038/nm.3127
|
| [117] |
Sun J, Liu Y, Tran J, et al. (2016) mTORC1-S6K1 inhibition or mTORC2 activation improves hippocampal synaptic plasticity and learning in Angelman syndrome mice. Cell Mol Life Sci 73: 4303–4314. doi: 10.1007/s00018-016-2269-z
|
| [118] |
van Slegtenhorst M, de Hoogt R, Hermans C, et al. (1997) Identification of the tuberous sclerosis gene TSC1 on chromosome 9q34. Science 277: 805–808. doi: 10.1126/science.277.5327.805
|
| [119] | European Chromosome 16 Tuberous Sclerosis Consortium (1993) Identification andcharacterization of the tuberous sclerosis gene on chromosome 16. Cell 75: 1305–1315. |
| [120] | Kwiatkowski DJ, Manning BD (2005) Tuberous sclerosis: a GAP at the crossroads of multiple signaling pathways. Hum Mol Genet 14 Spec No. 2: R251–R258. |
| [121] |
Hwang SK, Lee JH, Yang JE, et al. (2016) Everolimus improves neuropsychiatric symptoms in a patient with tuberous sclerosis carrying a novel TSC2 mutation. Mol Brain 9: 56. doi: 10.1186/s13041-016-0222-6
|
| [122] |
Meikle L, Pollizzi K, Egnor A, et al. (2008) Response of a neuronal model of tuberous sclerosis to mammalian target of rapamycin (mTOR) inhibitors: effects on mTORC1 and Akt signaling lead to improved survival and function. J Neurosci 28: 5422–5432. doi: 10.1523/JNEUROSCI.0955-08.2008
|
| [123] | Sato A, Kasai S, Kobayashi T, et al. (2012) Rapamycin reverses impaired social interaction in mouse models of tuberous sclerosis complex. Nat Commun 3: 1292. |
| [124] |
Williams VC, Lucas J, Babcock MA, et al., (2009) Neurofibromatosis type 1 revisited. Pediatrics 123: 124–133. doi: 10.1542/peds.2007-3204
|
| [125] | Garg S, Plasschaert E, Descheemaeker MJ, et al. (2015) Autism spectrum disorder profile in neurofibromatosis type I. J Autism Dev Disord 45: 1649–1657. |
| [126] | Plasschaert E, Descheemaeker MJ, Van Eylen L, et al. (2015) Prevalence of autism spectrum disorder symptoms in children with neurofibromatosis type 1. Am J Med Genet B Neuropsychiatr Genet 168B: 72–80. |
| [127] | Basu TN, Gutmann DH, Fletcher JA, et al. (1992) Aberrant regulation of ras proteins in malignant tumour cells from type 1 neurofibromatosis patients. Nature 356: 713–715. |
| [128] |
Guilding C, McNair K, Stone TW, et al. (2007) Restored plasticity in a mouse model of neurofibromatosis type 1 via inhibition of hyperactive ERK and CREB. Eur J Neurosci 25: 99–105. doi: 10.1111/j.1460-9568.2006.05238.x
|
| [129] |
Wang Y, Kim E, Wang X, et al. (2012) ERK inhibition rescues defects in fate specification of Nf1-deficient neural progenitors and brain abnormalities. Cell 150: 816–830. doi: 10.1016/j.cell.2012.06.034
|
| [130] | Acosta MT, Kardel PG, Walsh KS, et al. (2011) Lovastatin as treatment for neurocognitive deficits in neurofibromatosis type 1: phase I study. PediatrNeurol 45: 241–245. |
| [131] |
Li W, Cui Y, Kushner SA, et al. (2005) The HMG-CoA reductase inhibitor lovastatin reverses the learning and attention deficits in a mouse model of neurofibromatosis type 1. Curr Biol 15: 1961–1967. doi: 10.1016/j.cub.2005.09.043
|
| [132] |
Osterweil EK, Chuang SC, Chubykin AA, et al. (2013) Lovastatin corrects excess protein synthesis and prevents epileptogenesis in a mouse model of fragile X syndrome. Neuron 77: 243–250. doi: 10.1016/j.neuron.2012.01.034
|
| [133] | Waite KA, Eng C (2002) Protean PTEN: form and function. Am J Hum Genet 70: 829–844. |
| [134] |
Butler MG, Dasouki MJ, Zhou XP, et al. (2005) Subset of individuals with autism spectrum disorders and extreme macrocephaly associated with germline PTEN tumour suppressor gene mutations. J Med Genet 42: 318–321. doi: 10.1136/jmg.2004.024646
|
| [135] |
Goffin A, Hoefsloot LH, Bosgoed E, et al. (2001) PTEN mutation in a family with Cowden syndrome and autism. Am J Med Genet 105: 521–524. doi: 10.1002/ajmg.1477
|
| [136] | Maehama T, Dixon JE (1998) The tumor suppressor, PTEN/MMAC1, dephosphorylates the lipid second messenger, phosphatidylinositol 3,4,5-trisphosphate. J Biol Chem 273: 13375–13378. |
| [137] |
Penagarikano O, Mulle JG, Warren ST (2007) The pathophysiology of fragile X syndrome. Annu Rev Genomics Hum Genet 8: 109–129. doi: 10.1146/annurev.genom.8.080706.092249
|
| [138] | Brown V, Jin P, Ceman S, et al. (2001) Microarray identification of FMRP-associated brain mRNAs and altered mRNA translational profiles in fragile X syndrome. Cell 107: 477–487. |
| [139] | Darnell JC, Van Driesche SJ, Zhang C, et al. (2011) FMRP stalls ribosomal translocation on mRNAs linked to synaptic function and autism. Cell 146: 247–261. |
| [140] |
Gross C, Nakamoto M, Yao X, et al. (2010) Excess phosphoinositide 3-kinase subunit synthesis and activity as a novel therapeutic target in fragile X syndrome. J Neurosci 30: 10624–10638. doi: 10.1523/JNEUROSCI.0402-10.2010
|
| [141] |
Gross C, Chang CW, Kelly SM, et al (2015) Increased expression of the PI3K enhancer PIKE mediates deficits in synaptic plasticity and behavior in fragile X syndrome. Cell Rep 11: 727–736. doi: 10.1016/j.celrep.2015.03.060
|
| [142] | Darnell JC, Klann E (2013) The translation of translational control by FMRP: therapeutic targets for FXS. Nat Neurosci 16: 1530–1536. |
| [143] |
Richter JD, Bassell GJ, Klann E (2015) Deregulation and restoration of translational homeostasis in fragile X syndrome. Nat Rev Neurosci 16: 595–605. doi: 10.1038/nrn4001
|
| [144] |
Osterweil EK, Krueger DD, Reinhold K, et al. (2010) Hypersensitivity to mGluR5 and ERK1/2 leads to excessive protein synthesis in the hippocampus of a mouse model of fragile X syndrome. J Neurosci 30: 15616–15627. doi: 10.1523/JNEUROSCI.3888-10.2010
|
| [145] |
Tsai NP, Wilkerson JR, Guo W, et al. (2012) Multiple autism-linked genes mediate synapse elimination via proteasomal degradation of a synaptic scaffold PSD-95. Cell 151: 1581–1594. doi: 10.1016/j.cell.2012.11.040
|
| [146] |
Busquets-Garcia A, Maldonado R, Ozaita A (2014) New insights into the molecular pathophysiology of fragile X syndrome and therapeutic perspectives from the animal model. Int J Biochem Cell Biol 53: 121–126. doi: 10.1016/j.biocel.2014.05.004
|
| [147] | Berry-Kravis E, Des Portes V, Hagerman R, et al. (2016) Mavoglurant in fragile X syndrome: Results of two randomized, double-blind, placebo-controlled trials. Sci Transl Med 8: 321ra5. |
| [148] |
Shepherd GM, Katz DM (2011) Synaptic microcircuit dysfunction in genetic models of neurodevelopmental disorders: focus on Mecp2 and Met. Curr Opin Neurobiol 21: 827–833. doi: 10.1016/j.conb.2011.06.006
|
| [149] |
Lombardi LM, Baker SA, Zoghbi HY (2015) MECP2 disorders: from the clinic to mice and back. J Clin Invest 125: 2914–2923. doi: 10.1172/JCI78167
|
| [150] |
Chahrour M, Jung SY, Shaw C, et al. (2008) MeCP2, a key contributor to neurological disease, activates and represses transcription. Science 320: 1224–1229. doi: 10.1126/science.1153252
|
| [151] |
Jiang M, Ash RT, Baker SA, et al. (2013) Dendritic arborization and spine dynamics are abnormal in the mouse model of MECP2 duplication syndrome. J Neurosci 33: 19518–19533. doi: 10.1523/JNEUROSCI.1745-13.2013
|
| [152] |
Buiting K, Williams C, Horsthemke B (2016) Angelman syndrome-insights into a rare neurogenetic disorder. Nat Rev Neurol 12: 584–593. doi: 10.1038/nrneurol.2016.133
|
| [153] |
Kishino T, Lalande M, Wagstaff J (1997) UBE3A/E6-AP mutations cause Angelman syndrome. Nat Genet 15: 70–73. doi: 10.1038/ng0197-70
|
| [154] |
Vu TH, Hoffman AR (1997) Imprinting of the Angelman syndrome gene, UBE3A, is restricted to brain. Nat Genet 17: 12–13. doi: 10.1038/ng0997-12
|
| [155] |
Reiter LT, Seagroves TN, Bowers M, et al. (2006) Expression of the Rho-GEF Pbl/ECT2 is regulated by the UBE3A E3 ubiquitin ligase. Hum Mol Genet 15: 2825–2835. doi: 10.1093/hmg/ddl225
|
| [156] |
Jiang YH, Armstrong D, Albrecht U, et al. (1998) Mutation of the Angelman ubiquitin ligase in mice causes increased cytoplasmic p53 and deficits of contextual learning and long-term potentiation. Neuron 21: 799–811. doi: 10.1016/S0896-6273(00)80596-6
|
| [157] |
Mishra A, Godavarthi SK, Jana NR (2009) UBE3A/E6-AP regulates cell proliferation by promoting proteasomal degradation of p27. Neurobiol Dis 36: 26–34. doi: 10.1016/j.nbd.2009.06.010
|
| [158] |
Kumar S, Talis AL, Howley PM (1999) Identification of HHR23A as a substrate for E6-associated protein-mediated ubiquitination. J Biol Chem 274: 18785–18792. doi: 10.1074/jbc.274.26.18785
|
| [159] |
Greer PL, Hanayama R, Bloodgood BL, et al. (2010) The Angelman Syndrome protein Ube3A regulates synapse development by ubiquitinating arc. Cell 140: 704–716. doi: 10.1016/j.cell.2010.01.026
|
| [160] |
Margolis SS, Salogiannis J, Lipton DM, et al. (2010) EphB-mediated degradation of the RhoA GEF Ephexin5 relieves a developmental brake on excitatory synapse formation. Cell 143: 442–455. doi: 10.1016/j.cell.2010.09.038
|
| [161] |
Jay V, Becker LE, Chan FW, et al. (1991) Puppet-like syndrome of Angelman: a pathologic and neurochemical study. Neurology 41: 416–422. doi: 10.1212/WNL.41.3.416
|
| [162] |
Hethorn WR, Ciarlone SL, Filonova I, et al. (2015) Reelin supplementation recovers synaptic plasticity and cognitive deficits in a mouse model for Angelman syndrome. Eur J Neurosci 41: 1372–1380. doi: 10.1111/ejn.12893
|
| [163] |
Krishnan A, Zhang R, Yao V, et al. (2016) Genome-wide prediction and functional characterization of the genetic basis of autism spectrum disorder. Nat Neurosci 19: 1454–1462. doi: 10.1038/nn.4353
|
| [164] |
Kilpinen H, Ylisaukko-Oja T, Hennah W, et al. (2008) Association of DISC1 with autism and Asperger syndrome. Mol Psychiatry 13: 187–196. doi: 10.1038/sj.mp.4002031
|
| [165] |
Thomson PA, Parla JS, McRae AF, et al. (2014) 708 Common and 2010 rare DISC1 locus variants identified in 1542 subjects: analysis for association with psychiatric disorder and cognitive traits. Mol Psychiatry 19: 668–675. doi: 10.1038/mp.2013.68
|
| [166] |
Iossifov I, O'Roak BJ, Sanders SJ, et al. (2014) The contribution of de novo coding mutations to autism spectrum disorder. Nature 515: 216–221. doi: 10.1038/nature13908
|
| [167] |
Krumm N, Turner TN, Baker C, et al. (2015) Excess of rare, inherited truncating mutations in autism. Nat Genet 47: 582–588. doi: 10.1038/ng.3303
|
| [168] |
Brett M, McPherson J, Zang ZJ, et al. (2014) Massively parallel sequencing of patients with intellectual disability, congenital anomalies and/or autism spectrum disorders with a targeted gene panel. PLoS One 9: e93409. doi: 10.1371/journal.pone.0093409
|
| [169] |
Grønskov K, Brøndum-Nielsen K, Dedic A, et al. (2011) A nonsense mutation in FMR1 causing fragile X syndrome. Eur J Hum Genet 19: 489–491. doi: 10.1038/ejhg.2010.223
|
| [170] | Vincent JB, Konecki DS, Munstermann E, et al. (1996) Point mutation analysis of the FMR1 gene in autism. Mol Psychiatry 1: 227–231. |
| [171] |
Girirajan S, Dennis MY, Baker C, et al. (2013) Refinement and discovery of new hotspots of copy-number variation associated with autism spectrum disorder. Am J Hum Genet 92: 221–237. doi: 10.1016/j.ajhg.2012.12.016
|
| [172] |
Sebat J, Lakshmi B, Malhotra D, et al. (2007) Strong association of de novo copy number mutations with autism. Science 316: 445–449. doi: 10.1126/science.1138659
|
| [173] |
Zhao WW (2013) Intragenic deletion of RBFOX1 associated with neurodevelopmental/neuropsychiatric disorders and possibly other clinical presentations. Mol Cytogenet 6: 26. doi: 10.1186/1755-8166-6-26
|
| [174] |
Nguyen LS, Kim HG, Rosenfeld JA, et al. (2013) Contribution of copy number variants involving nonsense-mediated mRNA decay pathway genes to neuro-developmental disorders. Hum Mol Genet 22: 1816–1825. doi: 10.1093/hmg/ddt035
|
| [175] |
Talkowski ME, Rosenfeld JA, Blumenthal I, et al. (2012) Sequencing chromosomal abnormalities reveals neurodevelopmental loci that confer risk across diagnostic boundaries. Cell 149: 525–537. doi: 10.1016/j.cell.2012.03.028
|
| [176] |
Turner TN, Hormozdiari F, Duyzend MH, et al. (2016) Genome Sequencing of Autism-Affected Families Reveals Disruption of Putative Noncoding Regulatory DNA. Am J Hum Genet 98: 58–74. doi: 10.1016/j.ajhg.2015.11.023
|
| [177] |
Amir RE, Van den Veyver IB, Wan M, et al. (1999) Rett syndrome is caused by mutations in X-linked MECP2, encoding methyl-CpG-binding protein 2. Nat Genet 23: 185–188. doi: 10.1038/13810
|
| [178] | Hanchard NA, Carvalho CM, Bader P, et al. (2012) A partial MECP2 duplication in a mildly affected adult male: a putative role for the 3' untranslated region in the MECP2 duplication phenotype. BMC Med Genet 13: 71. |
| [179] |
Shibayama A, Cook EH Jr, Feng J, et al. (2004) MECP2 structural and 3'-UTR variants in schizophrenia, autism and other psychiatric diseases: a possible association with autism. Am J Med Genet B Neuropsychiatr Genet 128B: 50–53. doi: 10.1002/ajmg.b.30016
|
| [180] |
Helander A, Stödberg T, Jaeken J, et al. (2013) Dolichol kinase deficiency (DOLK-CDG) with a purely neurological presentation caused by a novel mutation. Mol Genet Metab 110: 342–344. doi: 10.1016/j.ymgme.2013.07.002
|
| [181] |
Epi4K Consortium, Epilepsy Phenome/Genome Project, Allen AS, et al. (2013) De novo mutations in epileptic encephalopathies. Nature 501: 217–221. doi: 10.1038/nature12439
|
| [182] |
McBride KL, Varga EA, Pastore MT, et al. (2010) Confirmation study of PTEN mutations among individuals with autism or developmental delays/mental retardation and macrocephaly. Autism Res 3: 137–141. doi: 10.1002/aur.132
|
| [183] |
Marui T, Hashimoto O, Nanba E, et al. (2004) Association between the neurofibromatosis-1 (NF1) locus and autism in the Japanese population. Am J Med Genet B Neuropsychiatr Genet 131B: 43–47. doi: 10.1002/ajmg.b.20119
|
| [184] |
Smalley SL (1998) Autism and tuberous sclerosis. J Autism Dev Disord 28: 407–414. doi: 10.1023/A:1026052421693
|
| [185] |
Serajee FJ, Nabi R, Zhong H, et al. (2003) Association of INPP1, PIK3CG, and TSC2 gene variants with autistic disorder: implications for phosphatidylinositol signalling in autism. J Med Genet 40: e119. doi: 10.1136/jmg.40.11.e119
|
| [186] |
Kong A, Frigge ML, Masson G, et al. (2012) Rate of de novo mutations and the importance of father's age to disease risk. Nature 488: 471–475. doi: 10.1038/nature11396
|
| [187] |
O'Roak BJ, Vives L, Girirajan S, et al. (2012) Sporadic autism exomes reveal a highly interconnected protein network of de novo mutations. Nature 485: 246–250. doi: 10.1038/nature10989
|
| [188] |
Harlalka GV, Baple EL, Cross H, et al. (2013) Mutation of HERC2 causes developmental delay with Angelman-like features. J Med Genet 50: 65–73. doi: 10.1136/jmedgenet-2012-101367
|
| [189] |
Puffenberger EG, Jinks RN, Wang H, et al. (2012) A homozygous missense mutation in HERC2 associated with global developmental delay and autism spectrum disorder. Hum Mutat 33: 1639–1646. doi: 10.1002/humu.22237
|
| [190] | Deciphering Developmental Disorders Study (2015) Large-scale discovery of novel genetic causes of developmental disorders. Nature 519: 223–228. |
| [191] |
Nava C, Lamari F, Héron D, et al. (2012) Analysis of the chromosome X exome in patients with autism spectrum disorders identified novel candidate genes, including TMLHE. Transl Psychiatry 2: e179. doi: 10.1038/tp.2012.102
|
| [192] |
Tastet J, Decalonne L, Marouillat S, et al. (2015) Mutation screening of the ubiquitin ligase gene RNF135 in French patients with autism. Psychiatr Genet 25: 263–267. doi: 10.1097/YPG.0000000000000100
|
| [193] |
Vourc'h P, Martin I, Bonnet-Brilhault F, et al. (2003) Mutation screening and association study of the UBE2H gene on chromosome 7q32 in autistic disorder. Psychiatr Genet 13: 221–225. doi: 10.1097/00041444-200312000-00005
|
| [194] | Noor A, Dupuis L, Mittal K, et al. (2015) 15q11.2 duplication encompassing only the UBE3A gene is associated with developmental delay and neuropsychiatric phenotypes. HumMutat 36: 689–693. |
| [195] |
Nurmi EL, Bradford Y, Chen Y, et al. (2001) Linkage disequilibrium at the Angelman syndrome gene UBE3A in autism families. Genomics 77: 105–113. doi: 10.1006/geno.2001.6617
|
| [196] |
Chahrour MH, Yu TW, Lim ET, et al. (2012) Whole-exome sequencing and homozygosity analysis implicate depolarization-regulated neuronal genes in autism. PLoS Genet 8: e1002635. doi: 10.1371/journal.pgen.1002635
|
| [197] |
Flex E, Ciolfi A, Caputo V, et al. (2013) Loss of function of the E3 ubiquitin-protein ligase UBE3B causes Kaufman oculocerebrofacial syndrome. J Med Genet 50: 493–499. doi: 10.1136/jmedgenet-2012-101405
|
| [198] |
Salyakina D, Cukier HN, Lee JM, et al. (2011) Copy number variants in extended autism spectrum disorder families reveal candidates potentially involved in autism risk. PLoS One 6: e26049. doi: 10.1371/journal.pone.0026049
|
| [199] | Kato T, Tamiya G, Koyama S, et al. (2012) UBR5 gene mutation is associated with familial adult myoclonic epilepsy in a Japanese family. ISRN Neurol 2012: 508308. |
| [200] |
Najmabadi H, Hu H, Garshasbi M, et al. (2011) Deep sequencing reveals 50 novel genes for recessive cognitive disorders. Nature 478: 57–63. doi: 10.1038/nature10423
|
| [201] |
Hao YH, Fountain MD Jr, FonTacer K, et al. (2015) USP7 acts as a molecular rheostat to promote WASH-dependent endosomal protein recycling and is mutated in a human neurodevelopmental disorder. Mol Cell 59: 956–969. doi: 10.1016/j.molcel.2015.07.033
|
| [202] | Wang K, Zhang H, Ma D, et al. (2009) Common genetic variants on 5p14.1 associate with autism spectrum disorders. Nature 459: 528–533. |
| [203] |
Piton A, Gauthier J, Hamdan FF, et al. (2011) Systematic resequencing of X-chromosome synaptic genes in autism spectrum disorder and schizophrenia. Mol Psychiatry 16: 867–880. doi: 10.1038/mp.2010.54
|
| [204] |
De Rubeis S, He X, Goldberg AP, et al. (2014) Synaptic, transcriptional and chromatin genes disrupted in autism. Nature 515: 209–215. doi: 10.1038/nature13772
|
| [205] |
Sanders SJ, He X, Willsey AJ, et al. (2015) Insights into autism spectrum disorder genomic architecture and biology from 71 Risk Loci. Neuron 87: 1215–1233. doi: 10.1016/j.neuron.2015.09.016
|
| [206] |
Lim ET, Raychaudhuri S, Sanders SJ, et al. (2013) Rare complete knockouts in humans: population distribution and significant role in autism spectrum disorders. Neuron 77: 235–242. doi: 10.1016/j.neuron.2012.12.029
|
| [207] |
Liao C, Fu F, Li R, et al. (2013) Loss-of-function variation in the DPP6 gene is associated with autosomal dominant microcephaly and mental retardation. Eur J Med Genet 56: 484–489. doi: 10.1016/j.ejmg.2013.06.008
|
| [208] |
Marshall CR, Noor A, Vincent JB, et al. (2008) Structural variation of chromosomes in autism spectrum disorder. Am J Hum Genet 82: 477–488. doi: 10.1016/j.ajhg.2007.12.009
|
| [209] |
Prontera P, Napolioni V, Ottaviani V, et al. (2014) DPP6 gene disruption in a family with Gilles de la Tourette syndrome. Neurogenetics 15: 237–242. doi: 10.1007/s10048-014-0418-9
|
| [210] |
Zhang Y, Manning BD (2015) mTORC1 signaling activates NRF1 to increase cellular proteasome levels. Cell Cycle 14: 2011–2017. doi: 10.1080/15384101.2015.1044188
|
| [211] |
Wang X, Proud CG (2006) The mTOR pathway in the control of protein synthesis. Physiology 21: 362–369. doi: 10.1152/physiol.00024.2006
|
| [212] |
Tilot AK, Frazier TW 2nd, Eng C (2015) Balancing proliferation and connectivity in PTEN-associated autism spectrum disorder. Neurotherapeutics 12: 609–619. doi: 10.1007/s13311-015-0356-8
|
| [213] |
Sharma A, Hoeffer CA, Takayasu Y, et al. (2010) Dysregulation of mTOR signaling in fragile X syndrome. J Neurosci 30: 694–702. doi: 10.1523/JNEUROSCI.3696-09.2010
|
| [214] |
Busquets-Garcia A, Gomis-González M, Guegan T, et al. (2013) Targeting the endocannabinoid system in the treatment of fragile X syndrome. Nat Med 19: 603–607. doi: 10.1038/nm.3127
|
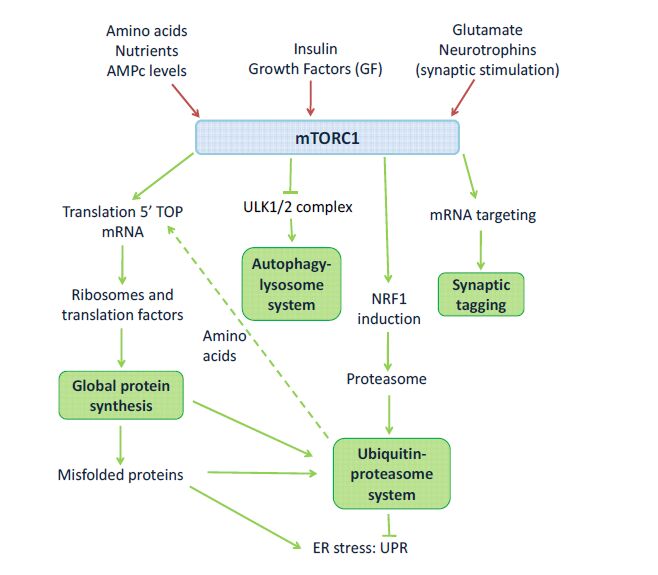
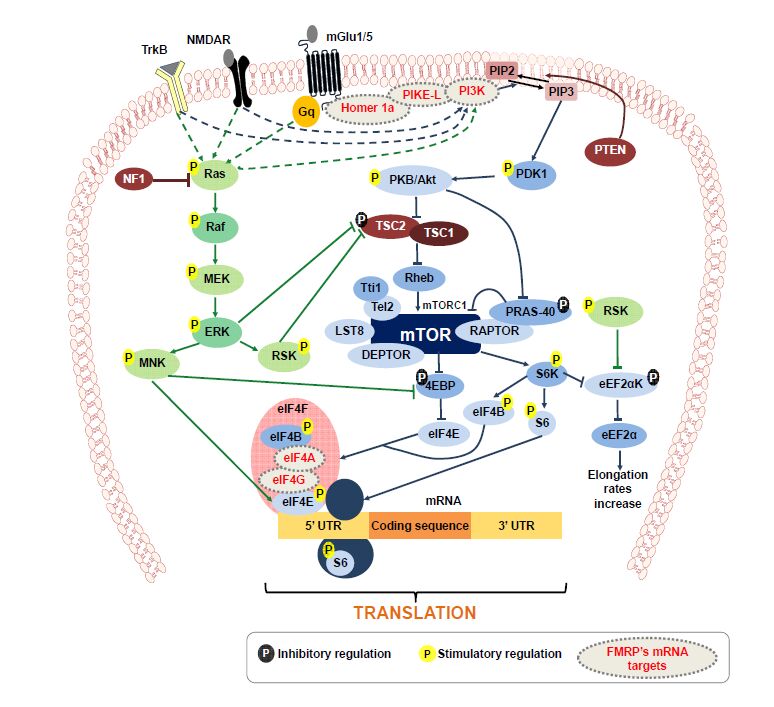
Figures(2) / Tables(2)

Judit Faus-Garriga, Isabel Novoa, Andrés Ozaita. mTOR signaling in proteostasis and its relevance to autism spectrum disorders[J]. AIMS Biophysics, 2017, 4(1): 63-89. doi: 10.3934/biophy.2017.1.63







 DownLoad:
DownLoad: