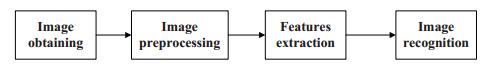
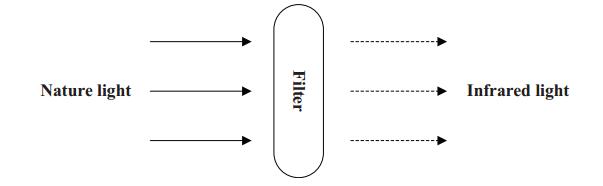
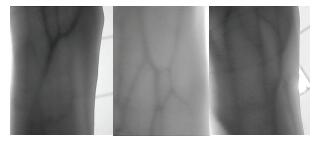
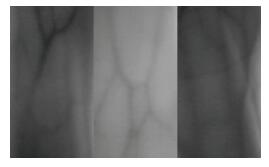
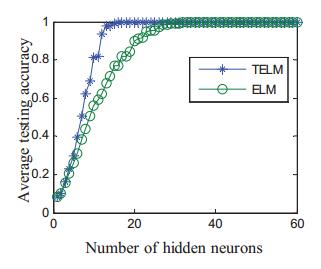
Vein recognition is a new identity authentication technology. It attracts many researchers' attention due to its good security and reliability. This paper proposes a wrist vein recognition system. The proposed system identifies people according to the characteristics of their wrist veins. A special camera is reformed to obtain the wrist vein images and an image dataset is established. Principal component analysis (PCA) is adopted to eliminate the redundant information in the images and extract their global features. The global features are classified by Two-hidden-layer Extreme Learning Machine (TELM). TELM is compared with original Extreme Learning Machine (ELM) and other two algorithms Support Vector Machine (SVM) and Naive Bayes (NB). Experiment results show that the accuracy of the proposed system is higher than the other three algorithms. Though the speed of TELM is not the fastest, it is able to recognize images within satisfactory time.
Citation: Cai-Tong Yue, Jing Liang, Bo-Fei Lang, Bo-Yang Qu. 2017: Two-hidden-layer extreme learning machine based wrist vein recognition system, Big Data and Information Analytics, 2(1): 59-68. doi: 10.3934/bdia.2017008
Vein recognition is a new identity authentication technology. It attracts many researchers' attention due to its good security and reliability. This paper proposes a wrist vein recognition system. The proposed system identifies people according to the characteristics of their wrist veins. A special camera is reformed to obtain the wrist vein images and an image dataset is established. Principal component analysis (PCA) is adopted to eliminate the redundant information in the images and extract their global features. The global features are classified by Two-hidden-layer Extreme Learning Machine (TELM). TELM is compared with original Extreme Learning Machine (ELM) and other two algorithms Support Vector Machine (SVM) and Naive Bayes (NB). Experiment results show that the accuracy of the proposed system is higher than the other three algorithms. Though the speed of TELM is not the fastest, it is able to recognize images within satisfactory time.

| [1] | Chen T., Cai J., Guo L. (2016) Research on hybrid face recognition algorithm based on voting Extreme Learning Machine. Journal of Zhengzhou University (Engineering Science) 37: 37-41. |
| [2] |
Cross J. M., Smith C. L. (1995) Thermographic imaging of the subcutaneous vascular network of the back of the hand for biometric identification. Institute of Electrical and Electronics Engineers Conference on Security Technology 20-35. doi: 10.1109/CCST.1995.524729

|
| [3] | Dong S., Yang J., Chen Y. (2015) Finger vein recognition based on multi-orientation weighted symmetric local graph structure. KSII Transactions on Internet and Information Systems 9: 4126-4142. |
| [4] |
Hashimoto J. (2006) Finger vein authentication technology and its future. Symposium on VLSI Circuits 5-8. doi: 10.1109/VLSIC.2006.1705285
|
| [5] |
Hsu C. B., Hao S. S., Lee J. C. (2011) Personal authentication through dorsal hand vein patterns. Optical Engineering 50: 087201, 10pp. doi: 10.1117/1.3607413
|
| [6] |
Huang G. B., Zhu Q. Y., Siew C. K. (2006) Extreme learning machine: Theory and applications. Neurocomputing 70: 489-501. doi: 10.1016/j.neucom.2005.12.126
|
| [7] | Im S. K., Park H. M., Kim Y. W. (2001) An biometric identification system by extracting hand vein patterns. Journal-korean Physical Society 3: 268-272. |
| [8] | Kono M., Ueki H., Umemura S. I. (2000) A new method for the identification of individuals by using of vein pattern matching of a finger. The Fifth Symposium on Pattern Mea-surement 9-12. |
| [9] |
Kumar A., Prathyusha K. V. (2009) Personal authentication using hand vein triangulation and knuckle shape. IEEE Transactions on Image processing 18: 2127-2136. doi: 10.1109/TIP.2009.2023153
|
| [10] |
Li X., Guo S., Gao F. (2007) Vein pattern recognitions by moment invariants. Bioinformatics and Biomedical Engineering 612-615. doi: 10.1109/ICBBE.2007.160
|
| [11] |
Mulyono D., Jinn H. S. (2008) A study of finger vein biometric for personal identification. International Symposium on Biometrics and Security Technologies 1-8. doi: 10.1109/ISBAST.2008.4547655
|
| [12] |
Qu B. Y., Lang B. F., Liang J. J. (2016) Two-hidden-layer extreme learning machine for regression and classification. Neurocomputing 175: 826-834. doi: 10.1016/j.neucom.2015.11.009
|
| [13] | Sugandhi N., Mathankumar M., Priya V. (2014) Real time authentication system using advanced finger vein recognition technique. International Conference on Communications and Signal Processing 1183-1187. |
| [14] | Wang J., Chang Q., Peng J. (2016) The Optimization of the Extreme Learning Machine and fitting analysis. Journal of Zhengzhou University (Engineering Science) 37: 20-24. |
| [15] |
Wang L., Leedham G. (2006) Near and far infrared imaging for vein pattern biometrics. Video and Signal Based Surveillance 52-58. doi: 10.1109/AVSS.2006.80
|
| [16] |
Wang Y., Li K., Cui J. (2010) Hand-dorsa vein recognition based on partition local binary pattern. IEEE 10th International Conference on Signal Processing 1671-1674. doi: 10.1109/ICOSP.2010.5656717
|
| [17] |
Wu K. S., Lee J. C., Lo T. M. (2013) A secure palm vein recognition system. Journal of Systems and Software 86: 2870-2876. doi: 10.1016/j.jss.2013.06.065
|
| [18] |
Yang J., Li X. (2010) Efficient finger vein localization and recognition. Pattern Recognition 1148-1151. doi: 10.1109/ICPR.2010.287
|
| [19] |
Yang W., Huang X., Zhou F. (2014) Comparative competitive coding for personal identification by using finger vein and finger dorsal texture fusion. Information Sciences 268: 20-32. doi: 10.1016/j.ins.2013.10.010
|
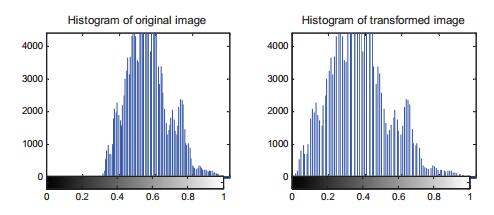
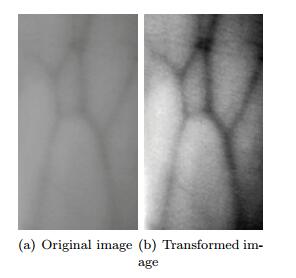
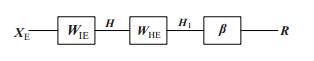
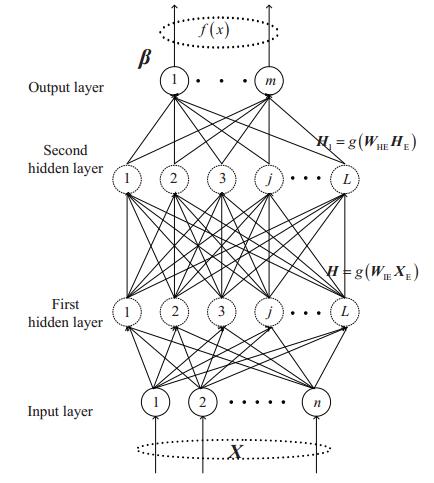
Figures(9) / Tables(2)

Cai-Tong Yue, Jing Liang, Bo-Fei Lang, Bo-Yang Qu. 2017: Two-hidden-layer extreme learning machine based wrist vein recognition system, Big Data and Information Analytics, 2(1): 59-68. doi: 10.3934/bdia.2017008









 DownLoad:
DownLoad: