Citation: Joseph Inungu, Kechi Iheduru-Anderson, Ossam J Odio. Recurrent Ebolavirus disease in the Democratic Republic of Congo: update and challenges[J]. AIMS Public Health, 2019, 6(4): 502-513. doi: 10.3934/publichealth.2019.4.502

| [1] |
Wannier SR, Worden L, Hoff NA, et al. (2019) Estimating the impact of violent events on transmission in Ebola virus disease outbreak, Democratic Republic of the Congo, 2018–2019. Epidemics 28: 100353. doi: 10.1016/j.epidem.2019.100353

|
| [2] | National Academies of Sciences, Engineering, and Medicine (2016) The Ebola Epidemic in West Africa: Proceedings of a Workshop. National Academies Press. |
| [3] |
Richardson JS, Dekker JD, Croyle MA, et al. (2010) Recent advances in Ebolavirus vaccine development. Hum Vaccines 6: 439–449. doi: 10.4161/hv.6.6.11097
|
| [4] |
Maxmen A (2019) Science under fire: Ebola researchers fight to test drugs and vaccines in a war zone. Nat 572: 16–17. doi: 10.1038/d41586-019-02258-4
|
| [5] | World Health Organization (2018) Notes for the record: consultation on Monitored Emergency Use of Unregistered and Investigational Interventions (MEURI) for Ebola virus disease (EVD). |
| [6] | Ilunga Kalenga O, Moeti M, Sparrow A, et al. (2019) The ongoing Ebola Epidemic in the Democratic Republic of Congo, 2018–2019. N Engl J Med. |
| [7] |
Kennedy SB, Bolay F, Kieh M, et al. (2017) Phase 2 placebo-controlled trial of two vaccines to prevent Ebola in Liberia. N Engl J Med 377: 1438–1447. doi: 10.1056/NEJMoa1614067
|
| [8] |
Sullivan NJ, Sanchez A, Rollin PE, et al. (2000) Development of a preventive vaccine for Ebola virus infection in primates. Nat 408: 605. doi: 10.1038/35046108
|
| [9] |
Geisbert TW, Pushko P, Anderson K, et al. (2002) Evaluation in nonhuman primates of vaccines against Ebola virus. Emerging Infect Dis 8: 503. doi: 10.3201/eid0805.010284
|
| [10] | World Health Organization (2019) Preliminary results on the efficacy of rVSV-ZEBOV- GP Ebola vaccine using the ring vaccination strategy in the control of an Ebola outbreak in the Democratic Republic of the Congo: an example of integration of research into epidemic response. Geneva: Organ. |
| [11] |
Baseler L, Chertow DS, Johnson KM, et al. (2017) The pathogenesis of Ebola virus disease. Annu Rev Pathol: Mech Dis 12: 387–418. doi: 10.1146/annurev-pathol-052016-100506
|
| [12] | World Health Organization (1978) Ebola haemorrhagic fever in Zaire, 1976. Report of an international commission. Bull World Health Organ 56: 271–293. |
| [13] | Rojek AM, Salam A, Ragotte RJ, et al. (2019) A systematic review and meta-analysis of patient data from the west Africa (2013–16) Ebola virus disease epidemic. Clin Microbiol Infect. |
| [14] | Benowitz I, Ackelsberg J, Balter SE, et al. (2014) Surveillance and preparedness for Ebola virus disease-New York City, 2014. MMWR Morbidity Mortal Wkly Rep 63: 934. |
| [15] |
Yuan J, Zhang Y, Li J, et al. (2012) Serological evidence of Ebolavirus infection in bats, China. Virol J 9: 236. doi: 10.1186/1743-422X-9-236
|
| [16] | Formenty P, Hatz C, Le Guenno B, et al. (1999) Human infection due to Ebola virus subtype Cote d'Ivoire: clinical and biologic presentation. J Infect Dis 179 (Supplement 1): S48–S53. |
| [17] |
Feldmann H (2014) Ebola-a growing threat? N Engl J Med 371: 1375–1378. doi: 10.1056/NEJMp1405314
|
| [18] |
Selvaraj SA, Lee KE, Harrell M, et al. (2018) Infection Rates and Risk Factors for Infection Among Health Workers During Ebola and Marburg Virus Outbreaks: A Systematic Review. J Infect Dis 218: S679–S689. doi: 10.1093/infdis/jiy435
|
| [19] |
Martínez MJ, Salim AM, Hurtado JC, et al. (2015) Ebola virus infection: overview and update on prevention and treatment. Infect Dis Ther 4: 365–390. doi: 10.1007/s40121-015-0079-5
|
| [20] |
Kaushik A, Tiwari S, Jayant RD, et al. (2016) Towards detection and diagnosis of Ebola virus disease at point-of-care. Biosens Bioelectron 75: 254–272. doi: 10.1016/j.bios.2015.08.040
|
| [21] |
Roddy P, Howard N, Van Kerkhove MD, et al. (2012) Clinical manifestations and case management of Ebola haemorrhagic fever caused by a newly identified virus strain, Bundibugyo, Uganda, 2007–2008. PloS One 7: e52986. doi: 10.1371/journal.pone.0052986
|
| [22] | Iwen PC, Smith PW, Hewlett AL, et al. (2015) Safety considerations in the laboratory testing of specimens suspected or known to contain Ebola virus. |
| [23] | Yang M, Ke Y, Liu C, et al. (2015) Diagnosis of Ebola virus disease: progress and prospects. Infect Dis Transl Med 1: 73–79. |
| [24] |
Saijo M, Niikura M, Morikawa S, et al. (2001) Immunofluorescence method for detection of Ebola virus immunoglobulin G, using HeLa cells which express recombinant nucleoprotein. J Clin Microbiol 39: 776–778. doi: 10.1128/JCM.39.2.776-778.2001
|
| [25] |
Li Y, Cu Y, Luo D (2005) Multiplexed detection of pathogen DNA with DNA-based fluorescence nanobarcodes. Nat Biotechnol 23: 885. doi: 10.1038/nbt1106
|
| [26] | Fenner F, Henderson DA, Arita I, et al. (1988) Smallpox and its eradication: World Health Organization Geneva. Available from: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC2491071/pdf/bullwho00076-0026.pdf. |
| [27] | World Health Organization (2014) Contact tracing during an outbreak of Ebola virus disease. |
| [28] | World Health Organization (2015) The ring vaccination trial: a novel cluster randomised controlled trial design to evaluate vaccine efficacy and effectiveness during outbreaks, with special reference to Ebola. BMJ Br Med J 351: h3740. |
| [29] | World Health Organization (2019) Ebola Virus Disease. Democratic Republic of the Congo External Situation Report 39. Available from: http://newsletters.afro.who.int/icfiles/1/46425/184054/6134450/97816cb57ede15249d4eb5b5/sit rep_evd_d%20rc_20190430-eng.pdf?ua=1,%20accessed%207%20May%202019. |
| [30] | Kadanali A, Karagoz G (2015) An overview of Ebola virus disease. North Clin Istanbul 2: 81. |
| [31] |
Marzi A, Robertson SJ, Haddock E, et al. (2015) VSV-EBOV rapidly protects macaques against infection with the 2014/15 Ebola virus outbreak strain. Sci 349: 739–742. doi: 10.1126/science.aab3920
|
| [32] |
Marzi A, Engelmann F, Feldmann F, et al. (2013) Antibodies are necessary for rVSV/ZEBOV-GP–mediated protection against lethal Ebola virus challenge in nonhuman primates. Proc Natl Acad Sci 110: 1893–1898. doi: 10.1073/pnas.1209591110
|
| [33] |
Pavot V (2016) Ebola virus vaccines: Where do we stand? Clin Immunol 173: 44–49. doi: 10.1016/j.clim.2016.10.016
|
| [34] |
Geisbert TW, Feldmann H (2011) Recombinant vesicular stomatitis virus–based vaccines against Ebola and Marburg virus infections. J Infect Dis 204: S1075–S1081. doi: 10.1093/infdis/jir349
|
| [35] | Roberts A, Buonocore L, Price R, et al. (1999) Attenuated vesicular stomatitis viruses as vaccine vectors. J Virol 73: 3723–3732. |
| [36] |
Rose NF, Marx PA, Luckay A, et al. (2001) An effective AIDS vaccine based on live attenuated vesicular stomatitis virus recombinants. Cell 106: 539–549. doi: 10.1016/S0092-8674(01)00482-2
|
| [37] |
Milligan ID, Gibani MM, Sewell R, et al. (2016) Safety and immunogenicity of novel adenovirus type 26–and modified vaccinia ankara–vectored ebola vaccines: a randomized clinical trial. Jama 315: 1610–1623. doi: 10.1001/jama.2016.4218
|
| [38] | Anywaine Z, Whitworth H, Kaleebu P, et al. (2019) Safety and Immunogenicity of a 2-Dose Heterologous Vaccination Regimen With Ad26.ZEBOV and MVA-BN-Filo Ebola Vaccines: 12-Month Data From a Phase 1 Randomized Clinical Trial in Uganda and Tanzania. J Infect Dis 220: 46–56. |
| [39] | Mutua G, Anzala O, Luhn K, et al. (2019) Safety and Immunogenicity of a 2-Dose Heterologous Vaccine Regimen With Ad26.ZEBOV and MVA-BN-Filo Ebola Vaccines: 12-Month Data From a Phase 1 Randomized Clinical Trial in Nairobi, Kenya. J Infect Dis 220: 57–67. |
| [40] | World Health Organization (2019) Ebola Vaccines Decision framework. Available from: https://www.who.int/blueprint/priority-%20diseases/keyaction/ebola-vaccinecandidates/en/,%20accessed%2007%20May%202019. |
| [41] |
Dhillon RS, Srikrishna D, Kelly JD (2018) Deploying RDTs in the DRC Ebola outbreak. Lancet 391: 2499–2500. doi: 10.1016/S0140-6736(18)31315-1
|
| [42] |
Fleck F (2009) The Democratic Republic of the Congo: quantifying the crisis. Bull World Health Organ 87: 6–7. doi: 10.2471/BLT.09.020109
|
| [43] | Vinck P, Pham PN, Bindu KK, et al. (2019) Institutional trust and misinformation in the response to the 2018–19 Ebola outbreak in North Kivu, DR Congo: a population-based survey. Lancet Infect Dis19: 529–536. |
| [44] |
Caron A, Bourgarel M, Cappelle J, et al. (2018) Ebola Virus Maintenance: If Not (Only) Bats, What Else? Viruses 10: 549. doi: 10.3390/v10100549
|
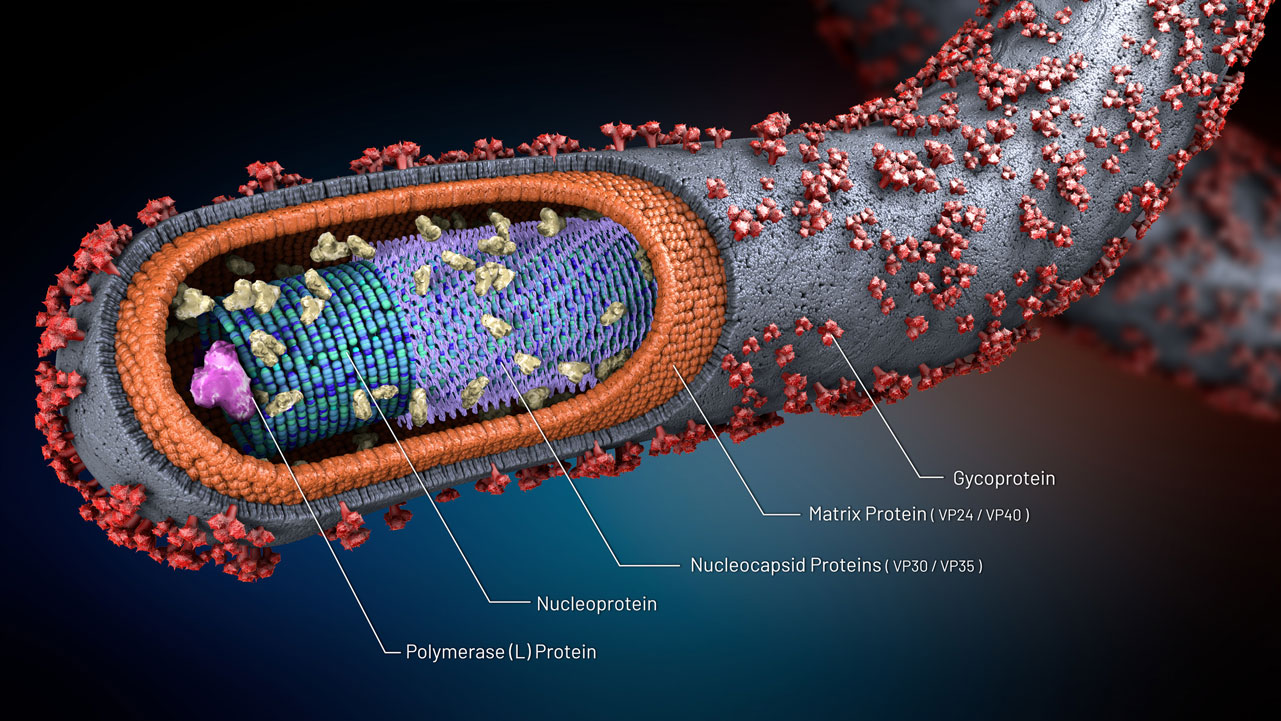
Figures(1) / Tables(1)

Joseph Inungu, Kechi Iheduru-Anderson, Ossam J Odio. Recurrent Ebolavirus disease in the Democratic Republic of Congo: update and challenges[J]. AIMS Public Health, 2019, 6(4): 502-513. doi: 10.3934/publichealth.2019.4.502







 DownLoad:
DownLoad: