Citation: Liang Wang, Zuobin Zhu, Huimin Qian, Ying Li, Ying Chen, Ping Ma, Bing Gu. Comparative genome analysis of 15 clinical Shigella flexneri strains regarding virulence and antibiotic resistance[J]. AIMS Microbiology, 2019, 5(3): 205-222. doi: 10.3934/microbiol.2019.3.205

| [1] |
Lima IFN, Havt A, Lima AAM (2015) Update on molecular epidemiology of Shigella infection. Cur Opin Gastroenterol 31: 30–37. doi: 10.1097/MOG.0000000000000136

|
| [2] |
Jennison AV, Verma NK (2004) Shigella flexneriinfection: pathogenesis and vaccine development. FEMS Microbiol Rev 28: 43–58. doi: 10.1016/j.femsre.2003.07.002
|
| [3] |
Kotloff KL, Riddle MS, Platts-Mills JA, et al. (2018) Shigellosis. The Lancet 391: 801–812. doi: 10.1016/S0140-6736(17)33296-8
|
| [4] |
Clements ACA, Thompson CN, Duy PT, et al. (2015) The rising dominance of Shigella sonnei: An intercontinental shift in the etiology of bacillary dysentery. PLOS Negl Trop Dis 9: e0003708. doi: 10.1371/journal.pntd.0003708
|
| [5] | Watson J, Jenkins C, Clements A, et al. (2018) Shigella sonnei does not use amoebae as protective hosts. Appl Environ Microb 84: e02679–17. |
| [6] | Ventola CL (2015) The antibiotic resistance crisis: part 1: causes and threats. P T 40: 277–283. |
| [7] |
Puzari M, Sharma M, Chetia P (2018) Emergence of antibiotic resistant Shigella species: A matter of concern. J Infect Public Health 11: 451–454. doi: 10.1016/j.jiph.2017.09.025
|
| [8] | Bhattacharya D, Sugunan AP, Bhattacharjee H, et al. (2012) Antimicrobial resistance in Shigella--rapid increase & widening of spectrum in Andaman Islands, India. Indian J Med Res 135: 365–370. |
| [9] | Schroeder GN, Hilbi H (2008) Molecular pathogenesis of Shigella spp.: Controlling host cell signaling, invasion, and death by Type III secretion. Clin Microbiol Rev 21: 134–156. |
| [10] | Bliven KA, Lampel KA (2017) Shigella: Virulence Factors and Pathogenicity. In: Gurtler JB, Doyle MP, Kornacki JL, Foodborne Pathogens, 7Eds., Springer, 169–208. |
| [11] |
Andersson DI, Hughes D (2010) Antibiotic resistance and its cost: is it possible to reverse resistance? Nat Rev Microbiol 8: 260–271. doi: 10.1038/nrmicro2319
|
| [12] | Schroeder M, Brooks B, Brooks A (2017) The complex relationship between virulence and antibiotic resistance. Genes 8: 1-23. |
| [13] |
Bonomo R, Geisinger E, Mortman NJ, et al. (2018) A global regulatory system links virulence and antibiotic resistance to envelope homeostasis in Acinetobacter baumannii. PLOS Pathog 14: e1007030c. doi: 10.1371/journal.ppat.1007030
|
| [14] |
Lee S, Hinz A, Bauerle E, et al. (2009) Targeting a bacterial stress response to enhance antibiotic action. Proc Nati Acad Sci 106: 14570–14575. doi: 10.1073/pnas.0903619106
|
| [15] | Roux D, Danilchanka O, Guillard T, et al. (2015) Fitness cost of antibiotic susceptibility during bacterial infection. Sci Transl Med 7: 297ra114–297ra114. |
| [16] |
Bankevich A, Nurk S, Antipov D, et al. (2012) SPAdes: A new genome assembly algorithm and its applications to single-cell sequencing. J Comput Biol 19: 455–477. doi: 10.1089/cmb.2012.0021
|
| [17] |
Darling ACE (2004) Mauve: Multiple alignment of conserved genomic sequence with rearrangements. Genome Res 14: 1394–1403. doi: 10.1101/gr.2289704
|
| [18] |
Edwards DJ, Holt KE (2013) Beginner's guide to comparative bacterial genome analysis using next-generation sequence data. Microb Inform Exp 3: 1-9. doi: 10.1186/2042-5783-3-1
|
| [19] |
Seemann T (2014) Prokka: rapid prokaryotic genome annotation. Bioinformatics 30: 2068–2069. doi: 10.1093/bioinformatics/btu153
|
| [20] |
Page AJ, Cummins CA, Hunt M, et al. (2015) Roary: rapid large-scale prokaryote pan genome analysis. Bioinformatics 31: 3691–3693. doi: 10.1093/bioinformatics/btv421
|
| [21] |
Alikhan N-F, Petty NK, Ben Zakour NL, et al. (2011) BLAST Ring Image Generator (BRIG): simple prokaryote genome comparisons. BMC Genomics 12: 402. doi: 10.1186/1471-2164-12-402
|
| [22] |
Price MN, Dehal PS, Arkin AP (2009) FastTree: computing large minimum evolution trees with profiles instead of a distance matrix. Mol Biol Evol 26: 1641–1650. doi: 10.1093/molbev/msp077
|
| [23] |
Letunic I, Bork P (2016) Interactive tree of life (iTOL) v3: an online tool for the display and annotation of phylogenetic and other trees. Nucleic Acids Res 44: W242–W245. doi: 10.1093/nar/gkw290
|
| [24] |
Chen L (2004) VFDB: a reference database for bacterial virulence factors. Nucleic Acids Res 33: D325–D328. doi: 10.1093/nar/gki008
|
| [25] | Johnson LS, Eddy SR, Portugaly E (2010) Hidden Markov model speed heuristic and iterative HMM search procedure. BMC Bioinformatics 11:1-8. |
| [26] | Abdelgader SA, Shi D, Chen M, et al. (2018) Antibiotics resistance genes screening and comparative genomics analysis of commensal Escherichia coli isolated from poultry farms between China and Sudan. BioMed Res Int 2018: 1–9. |
| [27] |
Projan SJ (2007) (Genome) Size Matters. Antimicrob Agents Ch 51: 1133–1134. doi: 10.1128/AAC.01370-06
|
| [28] |
Yang F (2005) Genome dynamics and diversity of Shigella species, the etiologic agents of bacillary dysentery. Nucleic Acids Res 33: 6445–6458. doi: 10.1093/nar/gki954
|
| [29] |
Beceiro A, Tomas M, Bou G (2013) Antimicrobial resistance and virulence: a successful or deleterious association in the bacterial world? Clin Microbiol Rev 26: 185–230. doi: 10.1128/CMR.00059-12
|
| [30] |
Parks AR, Peters JE (2009) Tn7 elements: Engendering diversity from chromosomes to episomes. Plasmid 61: 1–14. doi: 10.1016/j.plasmid.2008.09.008
|
| [31] |
Schulz zur Wiesch P, Engelstadter J, Bonhoeffer S (2010) Compensation of fitness costs and reversibility of antibiotic resistance mutations. Antimicrob Agents Ch 54: 2085–2095. doi: 10.1128/AAC.01460-09
|
| [32] |
Miskinyte M, Gordo I (2013) Increased survival of antibiotic-resistant Escherichia coli inside macrophages. Antimicrob Agents Ch 57: 189–195. doi: 10.1128/AAC.01632-12
|
| [33] |
Ramiro RS, Costa H, Gordo I (2016) Macrophage adaptation leads to parallel evolution of genetically diverse Escherichia coli small-colony variants with increased fitness in vivo and antibiotic collateral sensitivity. Evol Appl 9: 994–1004. doi: 10.1111/eva.12397
|
| [34] |
Cabral D, Wurster J, Belenky P (2018) Antibiotic persistence as a metabolic adaptation: stress, metabolism, the host, and new directions. Pharmaceuticals 11:1-19. doi: 10.3390/pharmaceutics11010001
|
| [35] | Pettibone GW, Sullivan SA, Shiaris MP (1987) Comparative survival of antibiotic-resistant and -sensitive fecal indicator bacteria in estuarine water. Appl Environ Microbiol 53: 1241–1245. |
| [36] |
Flint KP (1987) The long-term survival of Escherichia coliin river water. J Appl Bacteriol 63: 261–270. doi: 10.1111/j.1365-2672.1987.tb04945.x
|
 microbiol-05-03-205-s001.pdf microbiol-05-03-205-s001.pdf |
 |
| microbiol-05-03-205-s002.xlsx |
|
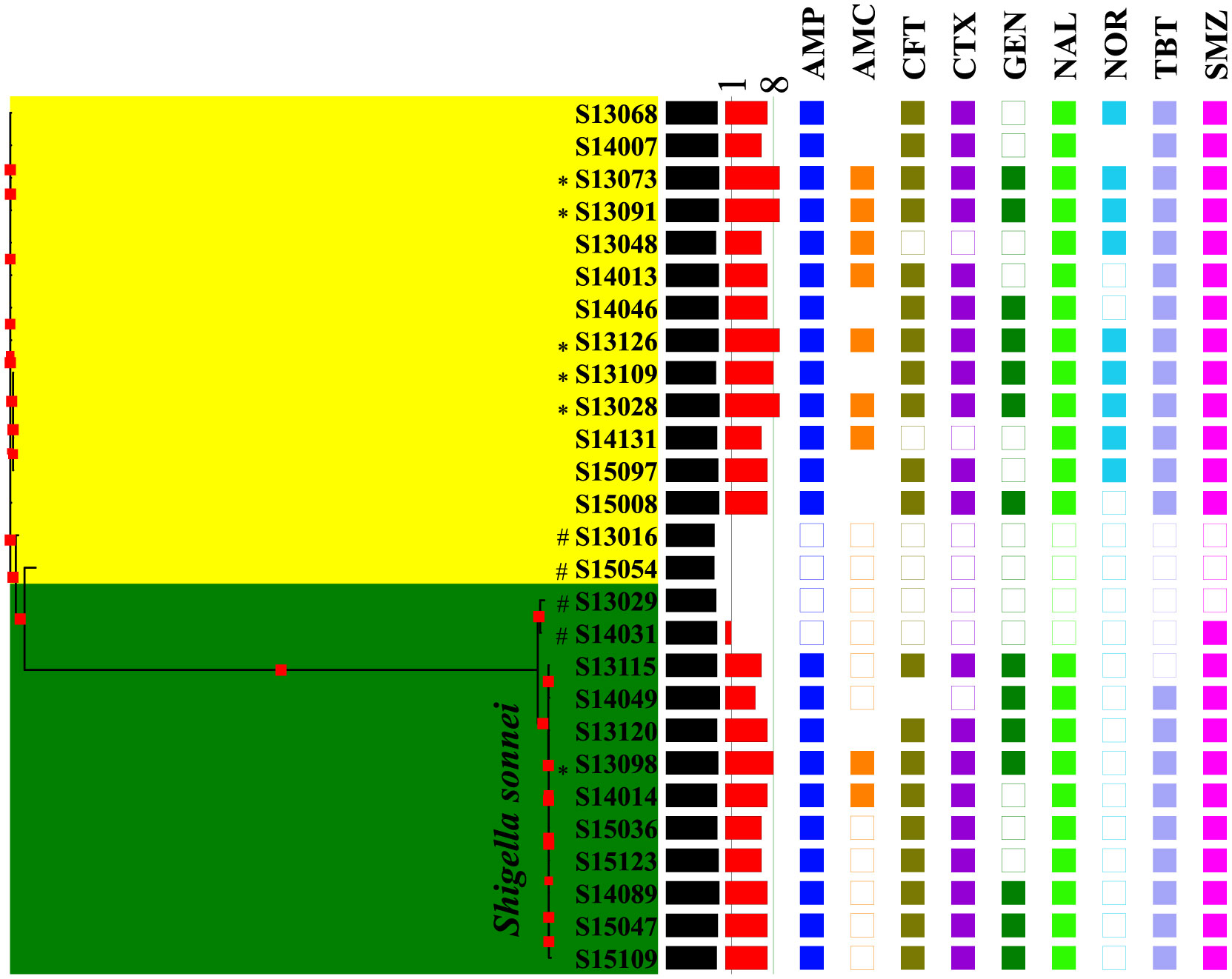
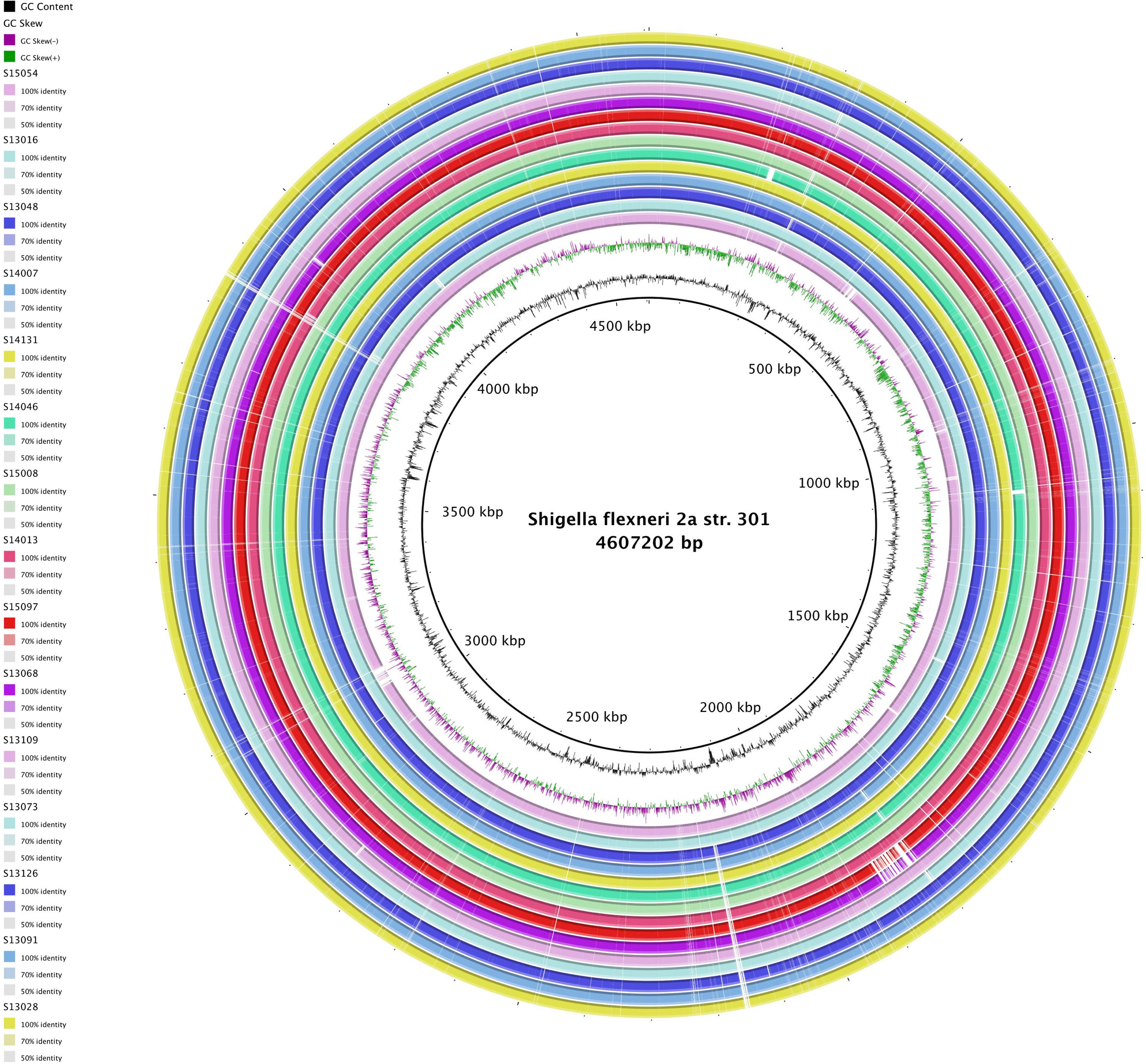
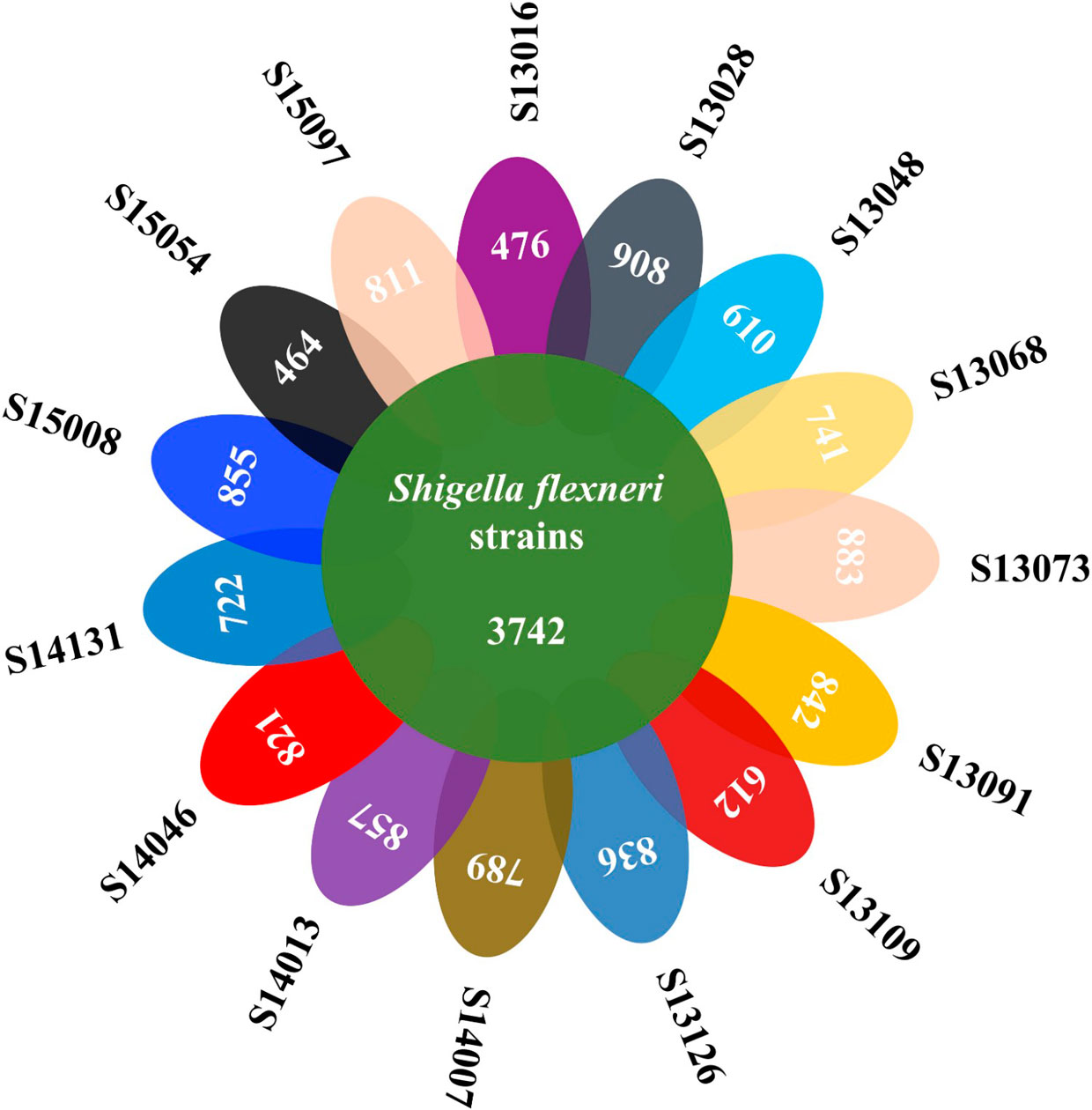
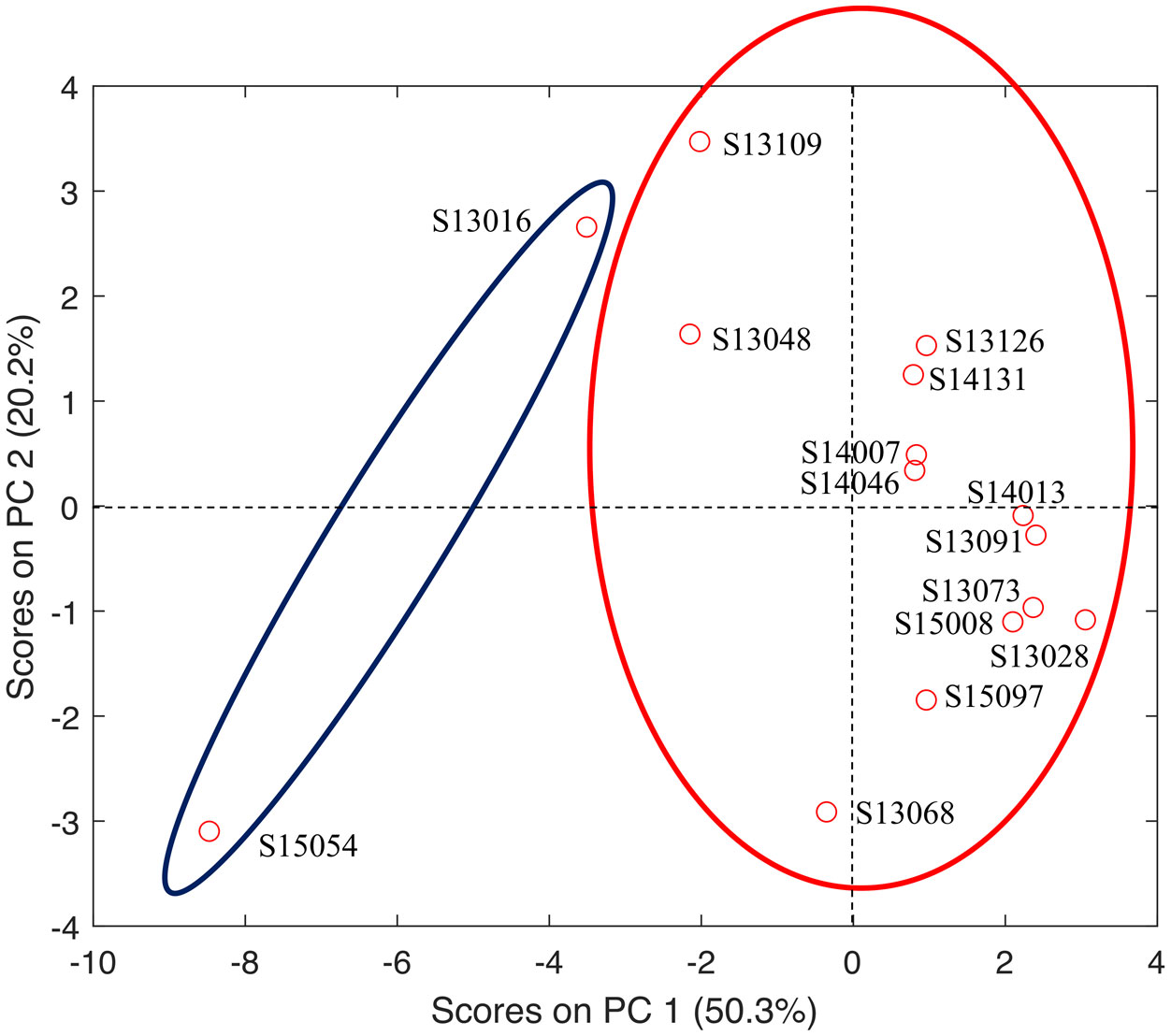
Figures(4) / Tables(3)

Liang Wang, Zuobin Zhu, Huimin Qian, Ying Li, Ying Chen, Ping Ma, Bing Gu. Comparative genome analysis of 15 clinical Shigella flexneri strains regarding virulence and antibiotic resistance[J]. AIMS Microbiology, 2019, 5(3): 205-222. doi: 10.3934/microbiol.2019.3.205







 DownLoad:
DownLoad: