Citation: Neetu Sharma, Sonu Bhatia, Abhinashi Singh Sodhi, Navneet Batra. Oral microbiome and health[J]. AIMS Microbiology, 2018, 4(1): 42-66. doi: 10.3934/microbiol.2018.1.42

| [1] | Berger G, Bitterman R, Azzam ZS (2015) The human microbiota: the rise of an "empire". Rambam Maimonides Med J 6: 1–5. |
| [2] |
Peek RM, Blaser MJ (2002) Helicobacter pylori and gastrointestinal tract adenocarcinomas. Nat Rev Cancer 2: 28–37. doi: 10.1038/nrc703

|
| [3] |
Human Microbiome Project Consortium (2012) Structure, function and diversity of the healthy human microbiome. Nature 486: 207–214. doi: 10.1038/nature11234
|
| [4] |
Charlson ES, Chen J, Custers-Allen R, et al. (2010) Disordered microbial communities in the upper respiratory tract of cigarette smokers. PLoS One 5: e15216. doi: 10.1371/journal.pone.0015216
|
| [5] |
Blaser MJ, Atherton JC (2004) Helicobacter pylori persistence: biology and disease. J Clin Invest 113: 321–333. doi: 10.1172/JCI20925
|
| [6] | Roberts AP, Kreth J (2014) The impact of horizontal gene transfer on the adaptive ability of the human oral microbiome. Front Cell Infect Mi 4: 124. |
| [7] |
Kort R, Caspers M, Graff A, et al. (2014) Shaping the oral microbiota through intimate kissing. Microbiome 2: 41. doi: 10.1186/2049-2618-2-41
|
| [8] | Willey JP (2008) Harley and Klein's Microbiology, In: Joanne MW, Linda MS, Christopher JW, 7 Eds., New York: McGraw-Hill Higher Education. |
| [9] | Wilson M (2008) The indigenous microbiota of the gastrointestinal tract, In: Wilson M, Author, Bacteriology of Humans: An Ecological Perspective, Oxford: Blackwell Publishing, 266–326. |
| [10] |
Miller MB, Bassler BL (2001) Quorum sensing in bacteria. Ann Rev Microbiol 55: 165–199. doi: 10.1146/annurev.micro.55.1.165
|
| [11] | Gerald PC (2013) Oral microbiome homeostasis: The new frontier in oral care therapies. J Dent Oral Disord Ther 1: 3. |
| [12] |
Joshi HM, Toleti RS (2009) Nutrition induced pleomorphism and budding mode of reproduction in Deinococcus radiodurans. BMC Res Notes 2: 123. doi: 10.1186/1756-0500-2-123
|
| [13] |
Dewhirst FE, Chen T, Izard J (2010) The human oral microbiome. J Bacteriol 192: 5002–5017. doi: 10.1128/JB.00542-10
|
| [14] |
Beck JD, Offenbacher S (2005) Systemic effects of periodontitis: epidemiology of periodontal disease and cardiovascular disease. J Periodontol 76: 2089–2100. doi: 10.1902/jop.2005.76.11-S.2089
|
| [15] |
Munson MA, Banerjee A, Watson TF, et al. (2004) Molecular analysis of the microflora associated with dental caries. J Clin Microbiol 42: 3023–3029. doi: 10.1128/JCM.42.7.3023-3029.2004
|
| [16] |
Genco RJ, Grossi SG, Ho A, et al. (2005) A proposed model linking inflammation to obesity, diabetes, and periodontal infections. J Periodontol 76: 2075–2084. doi: 10.1902/jop.2005.76.11-S.2075
|
| [17] |
Joshipura KJ, Hung HC, Rimm EB, et al. (2003) Periodontal disease, tooth loss, and incidence of ischemic stroke. Stroke 34: 47–52. doi: 10.1161/01.STR.0000052974.79428.0C
|
| [18] |
Awano S, Ansai T, Takata Y, et al. (2008) Oral health and mortality risk from pneumonia in the elderly. J Dent Res 87: 334–339. doi: 10.1177/154405910808700418
|
| [19] | Ford BJ, University of California Berkeley, Berkeley, 2008. Available from: http://www.ucmp.berkeley.eduhistoryleeuwenhoek.html. |
| [20] | Loesche WJ (1975) Chemotherapy of dental plaque infections. Oral Sci Rev 9: 65–107. |
| [21] | Wilson M (2005) Microbial inhabitants of humans: their ecology and role in health and disease, Cambridge: Cambridge University Press. |
| [22] | Alcaraz LD, Belda‐Ferre P, Cabrera‐Rubio R, et al. (2012) Identifying a healthy oral microbiome through metagenomics. Clin Microbiol Infec 18: 54–57. |
| [23] |
Avila M, Ojcius DM, Yilmaz O (2009) The oral microbiota: living with a permanent guest. DNA Cell Biol 28: 405–411. doi: 10.1089/dna.2009.0874
|
| [24] |
Peterson J, Garges S, Giovanni M, et al. (2009) The NIH Human microbiome project. Gen Res 19: 2317–2323. doi: 10.1101/gr.096651.109
|
| [25] | Mitreva M, Mardis ER (2009) Large-scale sequencing and analytical processing of ESTs, In: Parkinson J, Author, Expressed Sequence Tags (ESTs) Methods in Molecular Biology (Methods and Protocols), Humana Press, 153–187. |
| [26] |
Qin J, Li R, Raes J, et al. (2010) A human gut microbial gene catalogue established by metagenomic sequencing. Nature 464: 59–65. doi: 10.1038/nature08821
|
| [27] |
Kreth J, Merritt J, Shi W, et al. (2005) Competition and coexistence between Streptococcus mutans and Streptococcus sanguinis in the dental biofilm. J Bacteriol 187: 7193–7203. doi: 10.1128/JB.187.21.7193-7203.2005
|
| [28] |
Jenkinson HF, Lamont RJ (2005) Oral microbial communities in sickness and in health. Trends Microbiol 13: 589–595. doi: 10.1016/j.tim.2005.09.006
|
| [29] | Chalmers NI, Palmer RJ, Cisar JO, et al. (2008) Characterization of a Streptococcus sp.-Veillonella sp. community micromanipulated from dental plaque. J Bacteriol 190: 8145–8154. |
| [30] |
Kleinberg I (2002) A mixed-bacteria ecological approach to understanding the role of the oral bacteria in dental caries causation: an alternative to Streptococcus mutans and the specific-plaque hypothesis. Crit Rev Oral Biol Med 13: 108–125. doi: 10.1177/154411130201300202
|
| [31] |
Davey M, Otoole GA (2000) Microbial biofilms: from ecology to molecular genetics. Microbiol Mol Biol Rev 64: 847–867. doi: 10.1128/MMBR.64.4.847-867.2000
|
| [32] |
Kreft JU (2004) Biofilms promote altruism. Microbiology 150: 2751–2760. doi: 10.1099/mic.0.26829-0
|
| [33] | Irie Y, Parsek MR (2008) Quorum sensing and microbial biofilms. Curr Top Microbiol 322: 67–84. |
| [34] | Kolenbrander PE, Andersen RN, Blehert DS, et al. (2002) Communication among oral bacteria. Microbiol Mol Biol Rev 66: 486–505. |
| [35] |
Kuramitsu HK, He X, Lux R, et al. (2007) Interspecies interactions within oral microbial communities. Microbiol Mol Biol Rev 71: 653–670. doi: 10.1128/MMBR.00024-07
|
| [36] |
Kolenbrander PE (2000) Oral microbial communities: biofilms, interactions, and genetic systems. Annu Rev Microbiol 54: 413–437. doi: 10.1146/annurev.micro.54.1.413
|
| [37] |
McNab R, Ford SK, El-Sabaeny A, et al. (2003) LuxS-based signaling in Streptococcus gordonii: autoinducer 2 controls carbohydrate metabolism and biofilm formation with Porphyromonas gingivalis. J Bacteriol 185: 274–284. doi: 10.1128/JB.185.1.274-284.2003
|
| [38] |
Kreth J, Merritt J, Shi W, et al. (2005) Coordinated bacteriocin production and competence development: a possible mechanism for taking up DNA from neighbouring species. Mol Microbiol 57: 392–404. doi: 10.1111/j.1365-2958.2005.04695.x
|
| [39] |
van der Ploeg JR (2005) Regulation of bacteriocin production in Streptococcus mutans by the quorum-sensing system required for development of genetic competence. J Bacteriol 187: 3980–3989. doi: 10.1128/JB.187.12.3980-3989.2005
|
| [40] | Webb JS, Givskov M, Kjelleberg S (2003) Bacterial biofilms: prokaryotic adventures in multicellularity. Curr Opin Microbiol 6: 578–585. |
| [41] |
Antunes LCM, Ferreira RB, Buckner MM, et al. (2010) Quorum sensing in bacterial virulence. Microbiology 156: 2271–2282. doi: 10.1099/mic.0.038794-0
|
| [42] |
Jakubovics NS, Gill SR, Vickerman MM, et al. (2008) Role of hydrogen peroxide in competition and cooperation between Streptococcus gordonii and Actinomyces naeslundii. FEMS Microbiol Ecol 66: 637–644. doi: 10.1111/j.1574-6941.2008.00585.x
|
| [43] |
Edwards AM, Grossman TJ, Rudney JD (2006) Fusobacterium nucleatum transports noninvasive Streptococcus cristatus into human epithelial cells. Infect Immun 74: 654–662. doi: 10.1128/IAI.74.1.654-662.2006
|
| [44] |
Zhang G, Chen R, Rudney JD (2008) Streptococcus cristatus attenuates Fusobacterium nucleatum‐induced interleukin‐8 expression in oral epithelial cells. J Periodontal Res 43: 408–416. doi: 10.1111/j.1600-0765.2007.01057.x
|
| [45] | Yilmaz O (2008) The chronicles of Porphyromonas gingivalis: the microbium, the human oral epithelium and their interplay. Microbiology 154: 2897–2903. |
| [46] |
Bik EM, Long CD, Armitage GC, et al. (2010) Bacterial diversity in the oral cavity of 10 healthy individuals. ISME J 4: 962–974. doi: 10.1038/ismej.2010.30
|
| [47] |
Trim RD, Skinner MA, Farone MB, et al. (2011) Use of PCR to detect Entamoeba gingivalis in diseased gingival pockets and demonstrate its absence in healthy gingival sites. Parasitol Res 109: 857–864. doi: 10.1007/s00436-011-2312-9
|
| [48] |
Bahrani-Mougeot FK, Paster BJ, Coleman S, et al. (2008) Diverse and novel oral bacterial species in blood following dental procedures. J Clin Microbiol 46: 2129–2132. doi: 10.1128/JCM.02004-07
|
| [49] |
Ghannoum MA, Jurevic RJ, Mukherjee PK, et al. (2010) Characterization of the oral fungal microbiome (mycobiome) in healthy individuals. PLoS Pathog 6: e1000713. doi: 10.1371/journal.ppat.1000713
|
| [50] |
Wang J, Gao Y, Zhao F (2016) Phage-bacteria interaction network in human oral microbiome. Environ Microbiol 18: 2143–2158. doi: 10.1111/1462-2920.12923
|
| [51] | Baker JL, Bor B, Agnello M, et al. (2017) Ecology of the oral microbiome: beyond bacteria. Trends Microbiol 25: 362–374. |
| [52] |
Wahida A, Ritter K, Horz HP (2016) The Janus-Face of bacteriophages across human body habitats. PLoS Pathog 12: e1005634. doi: 10.1371/journal.ppat.1005634
|
| [53] |
Pride DT, Salzman J, Haynes M, et al. (2012) Evidence of a robust resident bacteriophage population revealed through analysis of the human salivary virome. ISME J 6: 915–926. doi: 10.1038/ismej.2011.169
|
| [54] | Ly M, Abeles SR, Boehm TK, et al. (2014) Altered oral viral ecology in association with periodontal disease. Mbio 5: e01133-14. |
| [55] | Arduino PG, Porter SR (2008) Herpes Simplex Virus Type 1 infection: overview on relevant clinic pathological features. J Oral Pathol Med 37: 107–121. |
| [56] | Wu YM, Yan J, Ojcius DM, et al. (2007) Correlation between infections with different genotypes of human cytomegalovirus and Epstein-Barr virus in subgingival samples and periodontal status of patients. J Clin Microbiol 45: 3665–3670. |
| [57] |
Giacaman RA, Asrani AC, Gebhard KH, et al. (2008) Porphyromonas gingivalis induces CCR5-dependent transfer of infectious HIV-1 from oral keratinocytes to permissive cells. Retrovirology 5: 1. doi: 10.1186/1742-4690-5-1
|
| [58] |
Herzberg MC, Weinberg A, Wahl SM (2006) The oral epithelial cell and first encounters with HIV-1. Adv Dent Res 19: 158–166. doi: 10.1177/154407370601900128
|
| [59] |
Loning T, Ikenberg H, Becker J, et al. (1985) Analysis of oral papillomas, leukoplakias, and invasive carcinomas for human papillomavirus type related DNA. J Invest Dermatol 84: 417–420. doi: 10.1111/1523-1747.ep12265517
|
| [60] | Kumaraswamy KL, Vidhya M (2011) Human papilloma virus and oral infections: an update. J Cancer Res 7: 120. |
| [61] |
Pride DT, Salzman J, Relman DA (2012) Comparisons of clustered regularly interspaced short palindromic repeats and viromes in human saliva reveal bacterial adaptations to salivary viruses. Environ Microbiol 14: 2564–2576. doi: 10.1111/j.1462-2920.2012.02775.x
|
| [62] |
Abeles SR, Robles-Sikisaka R, Ly M, et al. (2014) Human oral viruses are personal, persistent and gender-consistent. ISME J 8: 1753–1767. doi: 10.1038/ismej.2014.31
|
| [63] |
Stevens RH, Porras OD, Delisle AL (2009) Bacteriophages induced from lysogenic root canal isolates of Enterococcus faecalis. Oral Microbial Immun 24: 278–284. doi: 10.1111/j.1399-302X.2009.00506.x
|
| [64] |
Pride DT, Salzman J, Haynes M, et al. (2012) Evidence of a robust resident bacteriophage population revealed through analysis of the human salivary virome. ISME J 6: 915–926. doi: 10.1038/ismej.2011.169
|
| [65] |
Lepp PW, Brinig MM, Ouverney CC, et al. (2004) Methanogenic Archaea and human periodontal disease. P Natl Acad Sci USA 101: 6176–6181. doi: 10.1073/pnas.0308766101
|
| [66] |
Matarazzo F, Ribeiro AC, Feres M, et al. (2011) Diversity and quantitative analysis of Archaea in aggressive periodontitis and periodontally healthy subjects. J Clin Periodontol 38: 621–627. doi: 10.1111/j.1600-051X.2011.01734.x
|
| [67] |
Diaz PI, Chalmers NI, Rickard AH, et al. (2006) Molecular characterization of subject-specific oral microflora during initial colonization of enamel. Appl Environ Microb 72: 2837–2848. doi: 10.1128/AEM.72.4.2837-2848.2006
|
| [68] | Smillie CS, Smith MB, Friedman J, et al. (2011) Ecology drives a global network of gene exchange connecting the human microbiome. Nature 480: 241–244. |
| [69] |
Yaron S, Kolling GL, Simon L, et al. (2000) Vesicle-mediated transfer of virulence genes from Escherichia coli O157: H7 to other enteric vacteria. Appl Environ Microb 66: 4414–4420. doi: 10.1128/AEM.66.10.4414-4420.2000
|
| [70] |
Mashburn-Warren LM, Whiteley M (2006) Special delivery: vesicle trafficking in prokaryotes. Mol Microbiol 61: 839–846. doi: 10.1111/j.1365-2958.2006.05272.x
|
| [71] |
Warburton PJ, Palmer RM, Munson MA, et al. (2007) Demonstration of in vivo transfer of doxycycline resistance mediated by a novel transposon. J Antimicrob Chemo 60: 973–980. doi: 10.1093/jac/dkm331
|
| [72] |
Seville LA, Patterson AJ, Scott KP, et al. (2009) Distribution of tetracycline and erythromycin resistance genes among human oral and fecal metagenomic DNA. Microb Drug Resist 15: 159–166. doi: 10.1089/mdr.2009.0916
|
| [73] |
Ciric L, Brouwer MS, Mullany P (2014) Minocycline resistance in an oral Streptococcus infantis isolate is encoded by tet (S) on a novel small, low copy number plasmid. FEMS Microbiol Lett 353: 106–115. doi: 10.1111/1574-6968.12410
|
| [74] |
Connell SR, Tracz DM, Nierhaus KH (2003) Ribosomal protection proteins and their mechanism of tetracycline resistance. Antimicrob Agents Ch 47: 3675–3681. doi: 10.1128/AAC.47.12.3675-3681.2003
|
| [75] |
Nasidze I, Li J, Quinque D, et al. (2009) Global diversity in the human salivary microbiome. Gen Res 19: 636–643. doi: 10.1101/gr.084616.108
|
| [76] | Li Y, Ismail AI, Ge Y, et al. (2007) Similarity of bacterial populations in saliva from African-American mother-child dyads. J Clin Microbiol 45: 3082–3085. |
| [77] |
Corby PM, Bretz WA, Hart TC (2007) Heritability of oral microbial species in caries-active and caries-free twins. Twin Res Hum Genet 10: 821–828. doi: 10.1375/twin.10.6.821
|
| [78] | Edlund A, Santiago-Rodriguez TM, Boehm TK, et al. (2015) Bacteriophage and their potential roles in the human oral cavity. J Oral Microbial 7: 27423. |
| [79] | De Paepe M, Leclerc M, Tinsley CR, et al. (2014) Bacteriophages: an underestimated role in human and animal health? Front Cell Infect Mi 4: 39. |
| [80] | Dominguez-Bello MG, Costello EK, Contreras M, et al. (2010) Delivery mode shapes the acquisition and structure of the initial microbiota across multiple body habitats in newborns. P Natl Acad Sci USA 107: 11971–11975. |
| [81] | Holgerson PL, Harnevik L, Hernell O, et al. (2011) Mode of birth delivery affects oral microbiota in infants. J Dent Res 90: 1183–1188. |
| [82] |
Holgerson PL, Vestman NR, Claesson R, et al. (2013) Oral microbial profile discriminates breastfed from formula-fed infants. J Pediatr Gastr Nutr 56: 127. doi: 10.1097/MPG.0b013e31826f2bc6
|
| [83] |
Vestman NR, Timby N, Holgerson PL, et al. (2013) Characterization and in vitro properties of oral lactobacilli in breastfed infants. BMC Microbiol 13: 1. doi: 10.1186/1471-2180-13-1
|
| [84] | Baca P, Castillo AM, Liebana, MJ, et al. (2012) Horizontal transmission of Streptococcus mutans in school children. Med Oral Patol Oral 17: 495–500. |
| [85] | Stahringer SS, Clemente JC, Corley RP (2012) Nurture trumps nature in a longitudinal survey of salivary bacterial communities in twins from early adolescence to early adulthood. Genome Res 22: 2146–2152. |
| [86] | Brook I, Gober AE (2008) Recovery of potential pathogens in the nasopharynx of healthy and otitis media-prone children and their smoking and nonsmoking parents. Ann Otol Rhinol Laryngol 117: 727–730. |
| [87] | Arcavi L, Benowitz NL (2004) Cigarette smoking and infection. Arch Intern Med 164: 2206–2216. |
| [88] | Tamashiro E, Xiong G, Anselmo-Lima WT, et al. (2009) Cigarette smoke exposure impairs respiratory epithelial ciliogenesis. Am J Rhinol Allergy 23: 117–122. |
| [89] |
El Ahmer OR, Essery SD, Saadi AT, et al. (1999) The effect of cigarette smoke on adherence of respiratory pathogens to buccal epithelial cells. FEMS Immuno Med Microbiol 23: 27–36. doi: 10.1111/j.1574-695X.1999.tb01713.x
|
| [90] | Sapkota AR, Berger S, Vogel TM (2010) Human pathogens abundant in the bacterial metagenome of cigarettes. Environ Health Perspect 118: 351–356. |
| [91] |
Brook I, Gober AE (2005) Recovery of potential pathogens and interfering bacteria in the nasopharynx of smokers and nonsmokers. CHEST J 127: 2072–2075. doi: 10.1378/chest.127.6.2072
|
| [92] | Winkelhoff AV, Bosch-Tijhof CJ, Winkel EG, et al. (2001) Smoking affects the subgingival microflora in periodontitis. J Periodontol 72: 666–671. |
| [93] |
Fullmer SC, Preshaw PM, Heasman PA (2009) Smoking cessation alters subgingival microbial recolonization. J Dent Res 88: 524–528. doi: 10.1177/0022034509338676
|
| [94] |
Madhi SA, Adrian P, Kuwanda L, et al. (2007) Long-term effect of pneumococcal conjugate vaccine on nasopharyngeal colonization by Streptococcus pneumonia and associated interactions with Staphylococcus aureus and Haemophilus influenzae colonization in HIV-infected and HIV-uninfected children. J Infect Dis 196: 1662–1666. doi: 10.1086/522164
|
| [95] |
Regev-Yochay G, Trzciński K, Thompson CM, et al. (2006) Interference between Streptococcus pneumoniae and Staphylococcus aureus: in vitro hydrogen peroxide-mediated killing by Streptococcus pneumoniae. J Bacteriol 188: 4996–5001. doi: 10.1128/JB.00317-06
|
| [96] | McNally LM, Jeena PM, Gajee K, et al. (2006) Lack of association between the nasopharyngeal carriage of Streptococcus pneumoniae and Staphylococcus aureus in HIV-1-infected South African children. J Infect Dis 194: 385–390. |
| [97] |
Darveau RP (2010) Periodontitis: a polymicrobial disruption of host homeostasis. Nat Rev Microbiol 8: 481–490. doi: 10.1038/nrmicro2337
|
| [98] | Armitage GC (1999) Development of a classification system for periodontal diseases and conditions. Ann Periodontol 4: 1–6. |
| [99] | Socransky SS, Haffajee AD, Cugini MA, et al. (1998) Microbial complexes in subgingival plaque. J Clin Periodontol 25: 134–144. |
| [100] |
Kumar PS, Griffen AL, Barton JA, et al. (2003) New bacterial species associated with chronic periodontitis. J Dent Res 82: 338–344. doi: 10.1177/154405910308200503
|
| [101] |
Nyvad B, Kilian M (1987) Microbiology of the early colonization of human enamel and root surfaces in vivo. Eur J Oral Sci 95: 369–380. doi: 10.1111/j.1600-0722.1987.tb01627.x
|
| [102] | Zijnge V, van Leeuwen MBM, Degener JE, et al. (2010) Oral biofilm architecture on natural teeth. PLoS One 5: e9321. |
| [103] |
He XS, Shi WY (2009) Oral microbiology: past, present and future. Int J Oral Sci 1: 47. doi: 10.4248/ijos.09029
|
| [104] | Yamada T, Takahashi-Abbe S, Abbe K (1985) Effects of oxygen on pyruvate formate-lyase in situ and sugar metabolism of Streptococcus mutans and Streptococcus sanguis. Infect Immun 47: 129–134. |
| [105] | Takahashi N, Nyvad B (2011) The role of bacteria in the caries process ecological perspectives. J Dent Res 90: 294–303. |
| [106] | Downes J, Mantzourani M, Beighton D (2011) Scardovia wiggsiae sp. nov., isolated from the human oral cavity and clinical material, and emended descriptions of the genus Scardovia and Scardovia inopinata. Int J Syst Evol Microbiol 61: 25–29. |
| [107] | Kaur R, Gilbert SC, Sheehy EC, et al. (2013) Salivary levels of Bifidobacteria in caries free and caries active children. Int J Paediatr Dent 23: 32–38. |
| [108] | Munson MA, Pitt-Ford T, Chong B, et al. (2002) Molecular and cultural analysis of the microflora associated with endodontic infections. J Dent Res 81: 761–766. |
| [109] |
Burne RA, Marquis RE (2000) Alkali production by oral bacteria and protection against dental caries. FEMS Microbiol Lett 193: 1–6. doi: 10.1111/j.1574-6968.2000.tb09393.x
|
| [110] |
Razavi A, Gmür R, Imfeld T, et al. (2007) Recovery of Enterococcus faecalis from cheese in the oral cavity of healthy subjects. Oral Microbiol Immun 22: 248–251. doi: 10.1111/j.1399-302X.2006.00349.x
|
| [111] |
Kampfer J, Gohring TN, Attin T, et al. (2007) Leakage of food borne Enterococcus faecalis through temporary fillings in a simulated oral environment. Int Endontic J 40: 471–477. doi: 10.1111/j.1365-2591.2007.01252.x
|
| [112] | Raghavendran K, Mylotte JM, Scannapieco FA (2007) Nursing home‐associated pneumonia, hospital‐acquired pneumonia and ventilator‐associated pneumonia: the contribution of dental biofilms and periodontal inflammation. Periodontology 44: 164–177. |
| [113] |
Scannapieco FA (1999) Role of oral bacteria in respiratory infection. J Periodontol 70: 793–802. doi: 10.1902/jop.1999.70.7.793
|
| [114] |
Vincent JL (1999) Prevention of nosocomial bacterial pneumonia. Thorax 54: 544–549. doi: 10.1136/thx.54.6.544
|
| [115] | Gil-Perotin S, Ramirez P, Marti V, et al. (2012) Implications of endotracheal tube biofilm in ventilator-associated pneumonia response: a state of concept. Crit Care 16: 1–9. |
| [116] |
Marik PE (2001) Aspiration pneumonitis and aspiration pneumonia. Engl J Med 344: 665–671. doi: 10.1056/NEJM200103013440908
|
| [117] |
Scannapieco FA (2013) The oral microbiome: its role in health and in oral and systemic infections. Clin Microbiol Newsl 35: 163–169. doi: 10.1016/j.clinmicnews.2013.09.003
|
| [118] |
Borgnakke WS, Ostalo PV, Taylor GW, et al. (2013) Effect of periodontal disease on diabetes: systematic review of epidemiologic observational evidence. J Periodontol 84: 135–152. doi: 10.1902/jop.2013.132001
|
| [119] |
Segata N, Haake SK, Mannon P, et al. (2012) Composition of the adult digestive tract bacterial microbiome based on seven mouth surfaces, tonsils, throat and stool samples. Genome Biol 13: R42. doi: 10.1186/gb-2012-13-6-r42
|
| [120] |
Anand PS, Nandakumar K, Shenoy KT (2006) Are dental plaque, poor oral hygiene, and periodontal disease associated with Helicobacter pylori infection? J Periodontol 77: 692–698. doi: 10.1902/jop.2006.050163
|
| [121] | Whitmore SE, Lamont RJ (2014) Oral bacteria and cancer. PLoS Pathog 10: e1003933. |
| [122] |
Atanasova KR, Yilmaz O (2014) Looking in the Porphyromonas gingivalis cabinet of curiosities: the microbium, the host and cancer association. Mol Oral Microbiol 29: 55–66. doi: 10.1111/omi.12047
|
| [123] |
Ahn J, Chen CY, Hayes RB (2012) Oral microbiome and oral and gastrointestinal cancer risk. Cancer Cause Control 23: 399–404. doi: 10.1007/s10552-011-9892-7
|
| [124] |
Kaur S, White S, Bartold PM (2013) Periodontal disease and rheumatoid arthritis a systematic review. J Dent Res 92: 399–408. doi: 10.1177/0022034513483142
|
| [125] |
Kamer AR, Dasanayake AP, Craig RG, et al. (2008) Alzheimer's disease and peripheral infections: the possible contribution from periodontal infections, model and hypothesis. J Alzheimers Dis 13: 437–449. doi: 10.3233/JAD-2008-13408
|
| [126] | Ide M, Papapanou PN (2013) Epidemiology of association between maternal periodontal disease and adverse pregnancy outcomes-systematic review. J Clin Periodontol 40: S181–S194. |
| [127] | Zaura E, Keijser BJ, Huse SM, et al. (2009) Defining the healthy "core microbiome" of oral microbial communities. BMC Microbiol 9: 259. |
| [128] |
Lazarevic V, Whiteson K, Hernandez D, et al. (2010) Study of inter-and intra-individual variations in the salivary microbiota. BMC Genomics 11: 523. doi: 10.1186/1471-2164-11-523
|
| [129] |
Moon JH, Lee JH (2016) Probing the diversity of healthy oral microbiome with bioinformatics approaches. BMB reports 49: 662. doi: 10.5483/BMBRep.2016.49.12.164
|
| [130] | Baas-Becking LGM (1934) Geobiologie; of inleiding tot de milieukunde. WP Van Stockum & Zoon NV. |
| [131] | Loesche WJ (1969) Oxygen sensitivity of various anaerobic bacteria. Appl Microbiol 18: 723–727. |
| [132] | Ueki A, Akasaka H, Suzuki D, et al. (2006) Xylanibacter oryzae gen. nov., sp. nov., a novel strictly anaerobic, Gram negative, xylanolytic bacterium isolated from rice-plant residue in flooded rice-field soil in Japan. Int J Syst Evol Microbiol 56: 2215–2221. |
| [133] |
Ausec L, Kraighera B, Mandic-Mulec I (2009) Differences in the activity and bacterial community structure of drained grassland and forest peat soils. Soil Biol Biochem 41: 1874–1881. doi: 10.1016/j.soilbio.2009.06.010
|
| [134] |
Castro HF, Classen AT, Austin EE, et al. (2010) Soil microbial community responses to multiple experimental climate change drivers. Appl Environ Microb 76: 999–1007. doi: 10.1128/AEM.02874-09
|
| [135] |
Contreras M, Costello EK, Hidalgo G, et al. (2010) The bacterial microbiota in the oral mucosa of rural Amerindians. Microbiology 156: 3282–3287. doi: 10.1099/mic.0.043174-0
|
| [136] |
Li K, Bihan M, Methé BA (2013) Analyses of the stability and core taxonomic memberships of the Human microbiome. PLoS One 8: e63139. doi: 10.1371/journal.pone.0063139
|
| [137] | Zaura E, Nicu EA, Krom BP, et al. (2014) Acquiring and maintaining a normal oral microbiome: current perspective. Front Cell Infect Mi 4. |
| [138] | Galiminas V, Hall MW, Singh N, et al. (2014) Bacterial community composition of chronic periodontitis and novel oral sampling sites for detecting disease indicators. Microbiome 2: 32. |
| [139] | Sun B, Zhou D, Tu J, et al. (2017) Evaluation of the Bacterial Diversity in the Human Tongue Coating Based on Genus-Specific Primers for 16S rRNA Sequencing. Biomed Res Int 2017: 8184160. |
| [140] |
Hall MW, Singh N, Ng KF, et al. (2017) Inter-personal diversity and temporal dynamics of dental, tongue, and salivary microbiota in the healthy oral cavity. NPJ Biofilm Microbiome 3: 2. doi: 10.1038/s41522-016-0011-0
|
| [141] |
Takeshita T, Kageyama S, Furuta M, et al. (2016) Bacterial diversity in saliva and oral health-related conditions: the Hisayama Study. Sci Rep 6: 22164. doi: 10.1038/srep22164
|
| [142] |
Li J, Quinque D, Horz HP, et al. (2014) Comparative analysis of the human saliva microbiome from different climate zones: Alaska, Germany, and Africa. BMC Microbiol 14: 316. doi: 10.1186/s12866-014-0316-1
|
| [143] |
Huse SM, Ye Y, Zhou Y, et al. (2012) A Core human microbiome as viewed through 16S rRNA sequence clusters. PLoS One 7: e34242. doi: 10.1371/journal.pone.0034242
|
| [144] |
Gevers D, Knight R, Petrosino JF, et al. (2012) The human microbiome project: A community resource for the healthy human microbiome. PLoS Biol 10: e1001377. doi: 10.1371/journal.pbio.1001377
|
| [145] |
Walter J, Ley R (2011) The human gut microbiome: ecology and recent evolutionary changes. Annu Rev Microbiol 65: 411–429. doi: 10.1146/annurev-micro-090110-102830
|
| [146] |
Xiao E, Mattos M, Vieira GHA, et al. (2017) Diabetes enhances IL-17 expression and alters the oral microbiome to increase its pathogenicity. Cell Host Microbe 22: 120–128. doi: 10.1016/j.chom.2017.06.014
|
| [147] |
Grover HS, Blaggana A, Jain Y, et al (2015) Detection and measurement of oral malodor in chronic periodontitis patients and its correlation with levels of select oral anaerobes in subgingival plaque. Contemp Clin Dent 6: S181–S187. doi: 10.4103/0976-237X.166825
|
| [148] |
Hoceini A, Khelil NK, Ben-Yelles I, et al. (2016) Caries-related factors and bacterial composition of supragingival plaques in caries free and caries active Algerian adults. Asian Pac J Trop Biomed 6: 720–726. doi: 10.1016/j.apjtb.2016.06.011
|
| [149] | Horner-Devine MC, Lage M, Hughes JB, et al. (2004) A taxa-area relationship for bacteria. Nature 432: 750–753. |
| [150] |
Reid NM, Addison SL, Macdonald LJ, et al. (2011) Biodiversity of active and inactive bacteria in the gut flora of wood-feeding huhu beetle larvae (Prionoplus reticularis). Appl Environ Microb 77: 7000–7006. doi: 10.1128/AEM.05609-11
|
| [151] | Gong HL, Shi Y, Zhou L, et al. (2013) The composition of microbiome in larynx and the throat biodiversity between laryngeal squamous cell carcinoma patients and control population. PLoS One 8: e66476. |
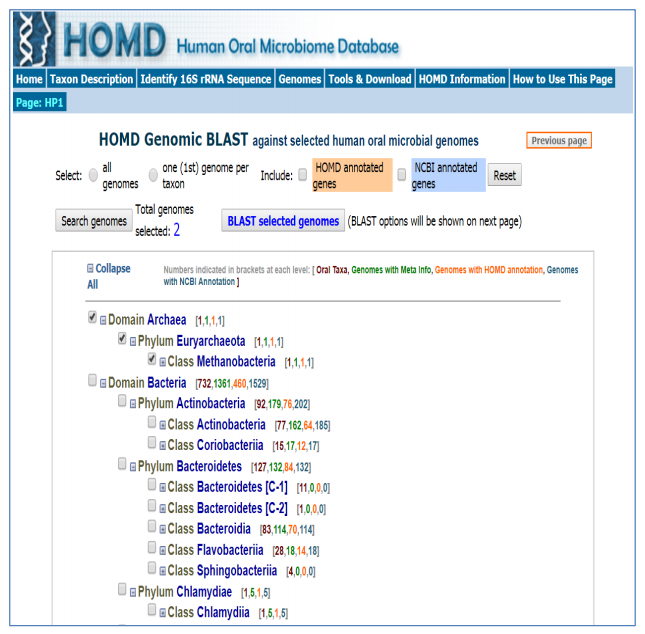
| [152] | Chen T, Yu WH, Izard J, et al. (2010) The Human Oral Microbiome Database: a web accessible resource for investigating oral microbe taxonomic and genomic information. Database 2010. |
| [153] | Human Microbiome Jumpstart Reference Strains Consortium (2010) A catalog of reference genomes from the human microbiome. Science 328: 994–999. |
| [154] |
Wang J, Qi J, Zhao H, et al. (2013) Metagenomic sequencing reveals microbiota and its functional potential associated with periodontal disease. Sci Rep 3: 1843. doi: 10.1038/srep01843
|
| [155] |
Raghunathan A, Ferguson HR, Bornarth CJ, et al. (2005) Genomic DNA amplification from a single bacterium. Appl Environ Microb 71: 3342–3347. doi: 10.1128/AEM.71.6.3342-3347.2005
|
| [156] | Dean FB, Hosono S, Fang L (2002) Comprehensive human genome amplification using multiple displacement amplification. P Natl Acad Sci USA 99: 5261–5266. |
| [157] |
Chitsaz H, Yee-Greenbaum JL, Tesler G (2011) Efficient de novo assembly of single-cell bacterial genomes from short-read data sets. Nat Biotechnol 29: 915–921. doi: 10.1038/nbt.1966
|
| [158] | Bankevich A, Nurk S, Antipov D, et al. (2012) SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J Comput Biol 19: 455–477. |
| [159] |
Lasken RS (2012) Genomic sequencing of uncultured microorganisms from single cells. Nat Rev Microbiol 10: 631–640. doi: 10.1038/nrmicro2857
|
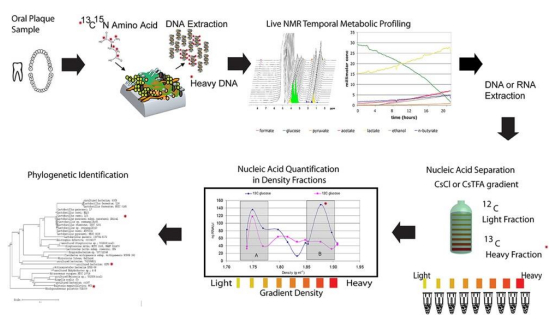
| [160] | Boschker HTS, Nold SC, Wellsbury P (1998) Direct linking of microbial populations to specific biogeochemical processes by 13C-labelling of biomarkers. Nature 392: 801–805. |
| [161] | Radajewski S, Ineson P, Parekh NR, et al. (2000) Stable-isotope probing as a tool in microbial ecology. Nature 403: 646–649. |
| [162] | McLean JS (2014) Advancements toward a systems level understanding of the human oral microbiome. Front Cell Infect Mi 4: 98. |
| [163] | Peterson SN, Snesrud E, Liu J, et al. (2013) The dental plaque microbiome in health and disease. PLoS One 8: e58487. |
| [164] |
Lourenço TGB, Heller D, Silva-Boghossian CM (2014) Microbial signature profiles of periodontally healthy and diseased patients. J Clin Periodontol 41: 1027–1036. doi: 10.1111/jcpe.12302
|
| [165] | Farrell JJ, Zhang L, Zhou H, et al. (2011) Variations of oral microbiota are associated with pancreatic diseases including pancreatic cancer. Gut 61: 582–588. |
| [166] |
Belda-Ferre P, Alcaraz LD, Cabrera-Rubio R, et al. (2012) The oral metagenome in health and disease. ISME J 6: 46–56. doi: 10.1038/ismej.2011.85
|
| [167] |
Duran-Pinedo AE, Chen T, Teles R, et al. (2014) Community-wide transcriptome of the oral microbiome in subjects with and without periodontitis. ISME J 8: 1659–1672. doi: 10.1038/ismej.2014.23
|
| [168] |
Jorth P, Trivedi U, Rumbaugh K, et al. (2013) Probing bacterial metabolism during infection using high-resolution transcriptomics. J Bacteriol 195: 4991–4998. doi: 10.1128/JB.00875-13
|
| [169] | Griffen AL, Beall CJ, Campbell JH, et al. (2012) Distinct and complex bacterial profiles in human periodontitis and health revealed by 16S pyrosequencing. ISME J 6: 1176–1185. |
| [170] | Schulze-Schweifing K, Banerjee A, Wade WG (2014).Comparison of bacterial culture and 16S rRNA community profiling by clonal analysis and pyrosequencing for the characterization of the dentine caries-associated microbiome. Front Cell Infect Mi 4: 164. |
| [171] |
Pushalkar S, Ji X, Li Y, et al. (2012) Comparison of oral microbiota in tumor and non-tumor tissues of patients with oral squamous cell carcinoma. BMC Microbiol 12: 144. doi: 10.1186/1471-2180-12-144
|
| [172] |
Liu B, Faller LL, Klitgord N, et al. (2012) Deep sequencing of the oral microbiome reveals signatures of periodontal disease. PLoS One 7: e37919. doi: 10.1371/journal.pone.0037919
|
| [173] | Xie G, Chain PSG, Lo CC, et al. (2010) Community and gene composition of a human dental plaque microbiota obtained by metagenomic sequencing. Mol Oral Microbiol 25: 391–405. |
| [174] |
Huson DH, Auch AF, Qi J, et al. (2007) MEGAN analysis of metagenomic data. Genome Res 17: 377–386. doi: 10.1101/gr.5969107
|
| [175] | Benitez-Paez A, Belda-Ferre P, Simon-Soro A, et al. (2014) Microbiota diversity and gene expression dynamics in human oral biofilms. BMC Genomics 15: 311. |
| [176] | Jorth P, Turner KH, Gumus P, et al. (2014) Metatranscriptomics of the human oral microbiome during health and disease. Mbio 5: e01012-14. |
| [177] |
Maron PA, Ranjard L, Mougel C, et al. (2007) Metaproteomics: a new approach for studying functional microbial ecology. Microb Ecol 53: 486–493. doi: 10.1007/s00248-006-9196-8
|
| [178] |
Yakob M, Fuentes L, Wang MB, et al. (2014) Salivary biomarkers for detection of oral squamous cell carcinoma: current state and recent advances. Curr Oral Health Rep 1: 133–141. doi: 10.1007/s40496-014-0014-y
|
| [179] |
Barnes VM, Ciancio SG, Shibly O, et al. (2011) Metabolomics reveals elevated macromolecular degradation in periodontal disease. J Dent Res 90: 1293–1297. doi: 10.1177/0022034511416240
|
| [180] | Arrais JP, Rosa N, Melo J, et al. (2013) Oral Card: a bioinformatic tool for the study of oral proteome. Arch Oral Biol 58: 762–772. |
Figures(3) / Tables(1)

Neetu Sharma, Sonu Bhatia, Abhinashi Singh Sodhi, Navneet Batra. Oral microbiome and health[J]. AIMS Microbiology, 2018, 4(1): 42-66. doi: 10.3934/microbiol.2018.1.42







 DownLoad:
DownLoad: