Citation: Mona Mohsen, Ehsan Gomaa, Nabila Ahmed Mazaid, Reem Mohammed. Synthesis and characterization of organic montmorillonite-polyvinyl alcohol-co-polyacrylic nanocomposite hydrogel for heavy metal uptake in water[J]. AIMS Materials Science, 2017, 4(5): 1122-1139. doi: 10.3934/matersci.2017.5.1122

| [1] | Abdel-Raouf MS, Abdul-Raheim ARM (2017) Removal of Heavy Metals from Industrial Waste Water by Biomass-Based Materials: A Review. J Pollut Eff Cont 5: 180. |
| [2] | Berihun D (2017) Removal of Chromium from Industrial Wastewater by Adsorption Using Coffee Husk. J Material Sci Eng 6: 331. |
| [3] |
Chen X, Zhou S, Zhang L, et al. (2016) Adsorption of Heavy Metals by Graphene Oxide/Cellulose Hydrogel Prepared from NaOH/Urea Aqueous Solution. Materials 9: 582. doi: 10.3390/ma9070582

|
| [4] |
Lingamdinne LP, Kim IS, Ha JH, et al. (2017) Enhanced Adsorption Removal of Pb(II) and Cr(III) by Using Nickel Ferrite-Reduced Graphene Oxide Nanocomposite. Metals 7: 225. doi: 10.3390/met7060225
|
| [5] |
Bajpai A, Tripathi N, Saxena S (2014) Nanocomposites Based on Natural Biodegradable Materials: Effect of Post-Preparative γ-Irradiation on the Swelling Properties. Open J Org Polym Mat 4: 10–15. doi: 10.4236/ojopm.2014.41003
|
| [6] |
Pandey N, Shukla SK, Singh NB (2017) Water purification by polymer nanocomposites: an overview. Nanocomposites 3: 47–66. doi: 10.1080/20550324.2017.1329983
|
| [7] |
El Adraa K, Georgelin T, Lambert JF, et al. (2017) Cysteine-montmorillonite composites for heavy metal cation complexation: A combined experimental and theoretical study. Chem Eng J 314: 406–417. doi: 10.1016/j.cej.2016.11.160
|
| [8] | Akpomie KG, Dawodu FA (2016) Acid-modified montmorillonite for sorption of heavy metals from automobile effluent. Beni-Seuf Univ J Basic Appl Sci 5: 1–12. |
| [9] |
Feng YL, Wang C, Mao N, et al. (2017) Research Progress of Organic Modified Montmorillonite. Adv Mater 6: 20–23. Available from: http://advinmaterials.org/article?journalid=129&doi=10.11648/j.am.20170603.11 doi: 10.11648/j.am.20170603.11
|
| [10] |
Burham N, Sayed M (2016) Adsorption Behavior of Cd2+ and Zn2+ onto Natural Egyptian Bentonitic Clay. Minerals 6: 129. doi: 10.3390/min6040129
|
| [11] | Rafiei HR, Shirvani M, Ogunseitan OA (2016) Removal of lead from aqueous solutions by a poly(acrylic acid)/bentonite nanocomposite. Appl Water Sci 6: 331–338. |
| [12] |
Ilgin P, Durak H, Gür A (2015) A Novel pH-Responsive p(AAm-co-METAC)/MMT Composite Hydrogel: Synthesis, Characterization and Its Absorption Performance on Heavy Metal Ions. Polym-Plast Tech Eng 54: 603–615. doi: 10.1080/03602559.2014.974189
|
| [13] | Mahaweero T, Thanatde J (2013) Extraction of Heavy Metals from Aqueous Solutions using Chitosan/Montmorillonite Hybrid Hydrogels [PhD Dissertation]. Case Western Reserve University. |
| [14] | Zhu L, Wang L, Xu Y (2017) Chitosan and surfactant co-modified montmorillonite: A multifunctional adsorbent for contaminant removal. Appl Clay Sci 146: 35–42. |
| [15] | Sirousazar M, Kokabi M, Hassan ZM, et al. (2011) Dehydration kinetics of polyvinyl alcohol nanocomposite hydrogels containing Na-montmorillonite nanoclay. Sci Iran 8: 780–784. |
| [16] | Maziad NA, Mohsen M, Gomaa E, et al. (2015) Radiation Copolymerization of Hydrogels Based in Polyacrylic Acid/Polyvinyl Alcohol Applied in Water Treatment Processes. J Mater Sci Eng A 5: 381–390. |
| [17] |
Kansy J (1996) Microcomputer program for analysis of positron annihilation lifetime spectra. Nucl Instrum Meth A 374: 235–244. doi: 10.1016/0168-9002(96)00075-7
|
| [18] |
Schmidt M, Maurer FHJ (2000) Relation between free-volume quantities from PVT–EOS analysis and PALS. Polymer 41: 8419–8424. doi: 10.1016/S0032-3861(00)00181-6
|
| [19] |
Jean Y (1990) Positron annihilation spectroscopy for chemical analysis: a novel probe for microstructural analysis of polymers. Microchem J 42: 72–102. doi: 10.1016/0026-265X(90)90027-3
|
| [20] |
Kokabi M, Sirousazar M, Hassan ZM (2007) PVA-Clay Nanocomposite Hydrogels for Wound Dressing. Eur Polym J 43: 773–781. doi: 10.1016/j.eurpolymj.2006.11.030
|
| [21] |
Barati A, Asgari M, Miri T, et al. (2013) Removal and Recovery of Copper and Nickel Ions from Aqueous Solution by Poly(Methacrylamide-Co-Acrylic Acid)/Montmorillonite Nanocomposites. Environ Sci Pollut R 20: 6242–6255. doi: 10.1007/s11356-013-1672-3
|
| [22] |
Yakar A (2014) Synthesis of Poly(N-Methylol Methacrylamide/Vinyl Sulfonic Acid) Hydrogels for Heavy Metal Ion Removal. B Korean Chem Soc 35: 3063–3070. doi: 10.5012/bkcs.2014.35.10.3063
|
| [23] |
El-Din HMN, Hegazy ESA, Ibraheim DM (2009) Synthesis and Characterization of Hydrogels Based on Gamma Irradiation of Acrylic Acid and Methacrylic Acid. Polym Composite 30: 569–575. doi: 10.1002/pc.20588
|
| [24] | Hegazy DE, Eid M, Madani M (2014) Effect of Ni Nano Particles on Thermal, Optical and Electrical Behaviour of Irradiated PVA/AAc Films. Arab J Nucl Sci Appl 47: 41–52. |
| [25] | Sapalidis AA, Katsaros FK, Kanellopoulos NK, et al. (2011) PVA/montmorillonite nanocomposites: development and properties, In: Cuppoletti J, Nanocomposites and polymers with analytical methods, InTech, 29–50. |
| [26] | Usuki A, Hasegawa N, Kato M, et al. (2005) Polymer-clay nanocomposites, In: Inorganic polymeric nanocomposites and membranes, Springer Berlin Heidelberg, 179: 135–195. |
| [27] | Părpăriţă E, Cheaburu CN, Pațachia SF, et al. (2014) Polyvinyl alcohol/chitosan/montmorillonite Nnanocomposites preparation by freeze/thaw cycles and characterization. Acta Chem Iasi 22: 75–96. |
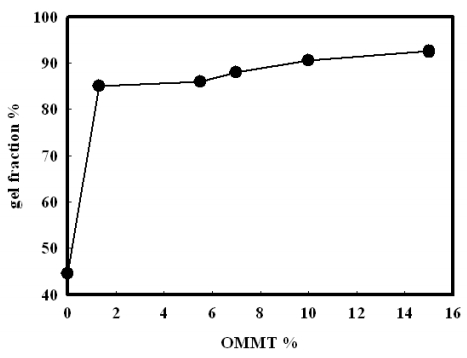
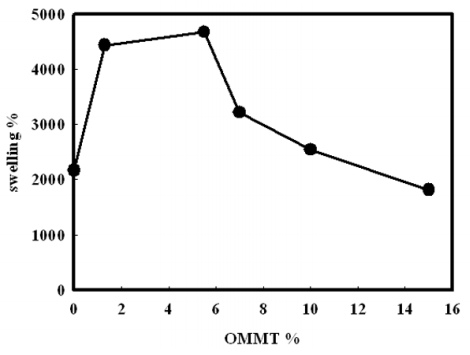
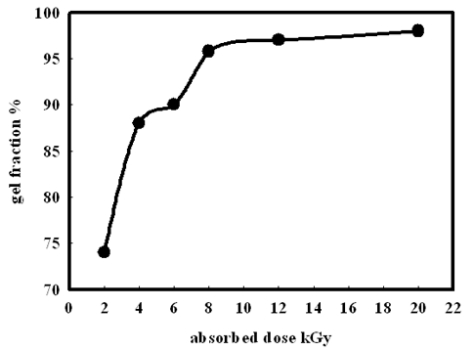
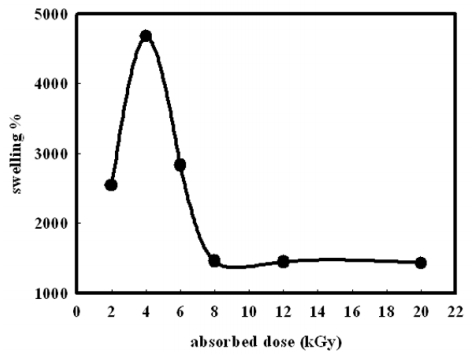
Figures(14) / Tables(2)

Mona Mohsen, Ehsan Gomaa, Nabila Ahmed Mazaid, Reem Mohammed. Synthesis and characterization of organic montmorillonite-polyvinyl alcohol-co-polyacrylic nanocomposite hydrogel for heavy metal uptake in water[J]. AIMS Materials Science, 2017, 4(5): 1122-1139. doi: 10.3934/matersci.2017.5.1122







 DownLoad:
DownLoad: