Citation: Tamara S. Wilson, Benjamin M. Sleeter, Jason Sherba, Dick Cameron. Land-use impacts on water resources and protected areas: applications of state-and-transition simulation modeling of future scenarios[J]. AIMS Environmental Science, 2015, 2(2): 282-301. doi: 10.3934/environsci.2015.2.282

| [1] | Turner BL, Clark WC, Kates RM, et al. (1993) The Earth as Transformed by Human Action: Global and Regional Changes in the Biosphere over the Past 300 Years. Cambridge University Press. |
| [2] |
Theobald DM (2010) Estimating natural landscape changes from 1992 to 2030 in the conterminous US. Landsc Ecol 25: 999-1011. doi: 10.1007/s10980-010-9484-z

|
| [3] |
Lewis DJ, Radeloff VC, Nelson E, et al. (2012) Economic-Based Projections of Future Land-Use Under Alternative Economic Policy Scenarios in the Conterminous US. Ecol Appl 22: 1036-1049. doi: 10.1890/11-0306.1
|
| [4] | U.S. Census Bureau (2013) State and County QuickFacts. U.S. Census Bureau. Available from: http://quickfacts.census.gov/qfd/index.html. |
| [5] | State of California (2013) State and County Total Population Projections, 2010-2060. State of California, Department of Finance. |
| [6] |
Tanaka SK, Zhu T, Lund JR, et al. (2006) Climate Warming and Water Management Adaptation for California. Clim Change 76: 361-387. doi: 10.1007/s10584-006-9079-5
|
| [7] |
Ault TR, Cole JE, Overpeck JT, et al. (2014) Assessing the Risk of Persistent Drought Using Climate Model Simulations and Paleoclimate Data. J Clim 27: 7529-7549. doi: 10.1175/JCLI-D-12-00282.1
|
| [8] |
Seabloom EW, Dobson AP, Stoms DM (2002) Extinction rates under nonrandom patterns of habitat loss. Proc Natl Acad Sci U S A 99: 11229-11234. doi: 10.1073/pnas.162064899
|
| [9] |
Davies RG, Orme CDL, Olson V, et al. (2006) Human impacts and the global distribution of extinction risk. Proc R Soc B 273: 2127-2133. doi: 10.1098/rspb.2006.3551
|
| [10] |
Rittenhouse CD, Pidgeon AM, Albright TP, et al. (2012) Land-Cover Change and Avian Diversity in the Conterminous United States: Land-Cover Change and Avian Diversity. Conserv Biol 26: 821-829. doi: 10.1111/j.1523-1739.2012.01867.x
|
| [11] |
Ross Z, English PB, Scalf R, et al. (2006) Nitrogen dioxide prediction in Southern California using land use regression modeling: potential for environmental health analyses. J Expo Sci Environ Epidemiol 16: 106-114. doi: 10.1038/sj.jea.7500442
|
| [12] |
Foley JA (2005) Global Consequences of Land Use. Science 309: 570-574. doi: 10.1126/science.1111772
|
| [13] | Houghton RA, Hackler JL (2008) Carbon Flux to the Atmosphere from Land-Use Changes 1850-2005. Carbon Dioxide Information Analysis Center, Oak Ridge National Laboratory, ORNL/CDIAC-131 NDP-050/R1, Oak Ridge, Tennessee. |
| [14] |
Lawrence PJ, Chase TN (2010) Investigating the climate impacts of global land cover change in the community climate system model. Int J Climatol 30: 2066-2087. doi: 10.1002/joc.2061
|
| [15] |
Pitman AJ, Avila FB, Abramowitz G, et al. (2011) Importance of background climate in determining impact of land-cover change on regional climate. Nat Clim Change 1: 472-475. doi: 10.1038/nclimate1294
|
| [16] |
Dale VH (1997) The relationship between land-use change and climate change. Ecol Appl 7: 753-769. doi: 10.1890/1051-0761(1997)007[0753:TRBLUC]2.0.CO;2
|
| [17] |
Sala OE (2000) Global Biodiversity Scenarios for the Year 2100. Science 287: 1770-1774. doi: 10.1126/science.287.5459.1770
|
| [18] |
Orang MN, Matyac S, Snyder RL (2008) Survey of Irrigation Methods in California in 2001. J Irrig Drain Eng 134: 96-100. doi: 10.1061/(ASCE)0733-9437(2008)134:1(96)
|
| [19] | U.S. Department of Agriculture (1994) 1992 Census of Agiculture: California, state and county data. U.S. Department of Commerce, Economics and Statistics Administration, Bureau of the Census. |
| [20] | U.S. Department of Agriculture (1999) 1997 Census of Agiculture: California, state and county data. U.S. Department of Agriculture, National Agricultural Statistics Service. |
| [21] | U.S. Department of Agriculture (2004) 2002 Census of Agiculture: California, state and county data. U.S. Department of Agriculture, National Agricultural Statistics Service. |
| [22] | U.S. Department of Agriculture (2009) 2007 Census of Agiculture: California, state and county data. U.S. Department of Agriculture, National Agricultural Statistics Service. |
| [23] | U.S. Department of Agriculture (2014) 2012 Census of Agiculture: California, state and county data. U.S. Department of Agriculture, National Agricultural Statistics Service. |
| [24] |
Tanaka SK, Zhu T, Lund JR, et al. (2006) Climate Warming and Water Management Adaptation for California. Clim Chang 76: 361-387. doi: 10.1007/s10584-006-9079-5
|
| [25] |
Margules CR, Pressey RL (2000) Systematic conservation planning. Nature 405: 243-253. doi: 10.1038/35012251
|
| [26] |
Wilson TS, Sleeter BM, Sleeter RR, et al. (2014) Land-Use Threats and Protected Areas: A Scenario-Based, Landscape Level Approach. Land 3: 362-389. doi: 10.3390/land3020362
|
| [27] | Wilson TS, Sleeter BM, Davis AW (2014) Potential future land use threats to California's protected areas. Reg Environ Change 1-14. |
| [28] | Piekielek NB, Hansen AJ (2012) Extent of fragmentation of coarse-scale habitats in and around U.S. Neational Parks. Biol Conserv 155: 13-22. |
| [29] | Radeloff VC, Stewart SI, Hawbaker TJ, et al. (2009) Housing growth in and near United States protected areas limits their conservation value. Proc Natl Acad Sci 107:940-945. |
| [30] |
Fleishman E, Blockstein DE, Hall JA, et al. (2011) Top 40 Priorities for Science to Inform US Conservation and Management Policy. BioScience 61: 290-300. doi: 10.1525/bio.2011.61.4.9
|
| [31] | Ricketts T, Imhoff M (2003) Biodiversity, urban areas, and agriculture: locating priority ecoregions for conservation. Conserv Ecol 8: 1-15. |
| [32] | U.S. Environmental Protection Agency (2013) Level III ecoregions of the continental United States, Digital map. http://www.epa.gov/wed/pages/ecoregions/level_iii_iv.htm. |
| [33] |
Omernik JM (1987) Ecoregions of the conterminous United States. Ann Assoc Am Geograghers 77: 118-125. doi: 10.1111/j.1467-8306.1987.tb00149.x
|
| [34] |
Gallant AL, Loveland TR, Sohl TL (2004) Using a geographic framework for analyzing land cover issues. Environ Manage 34: 89-110. doi: 10.1007/s00267-003-0145-3
|
| [35] | Vogelmann JE, Howard SM, Yang L, et al. (2001) Completion of the 1990's national land cover data set for the conterminous United States. Photogramm Eng Remote Sens 61: 650-662. |
| [36] | Protected Areas Database of the United States (PAD-US) (2012) version 1.3 Combined Feature Class. Available from: http://gapanalysis.usgs.gov/padus/. |
| [37] | U.S. Department of Agriculture (2010) National Agricultural Statistics Service, 2010 California Cropland Data Layer. Available from: http://www.nass.usda.gov/research/Cropland/metadata/metadata_ca10.htm. |
| [38] | California Department of Conservation (2014) Farmland Mapping and Monitoring Program. Available from: ftp://ftp.consrv.ca.gov/pub/dlrp/FMMP/. |
| [39] | Nakicenovic N, Swart R (2000) IPCC Special Report on Emission Scenarios. Cambridge University Press, Cambridge, UK. |
| [40] | Heistermann M, Müller C, Ronneberger K (2006) Land in sight? Achievements, deficits and potentials of continental to global scale land-use modeling. Agric Ecosyst Environ 114: 141-158. |
| [41] |
Rounsevell MDA, Reginster I, Araújo MB, et al. (2006) A coherent set of future land use change scenarios for Europe. Agric Ecosyst Environ 114: 57-68. doi: 10.1016/j.agee.2005.11.027
|
| [42] |
Sleeter BM, Sohl TL, Bouchard MA, et al. (2012) Scenarios of land use and land cover change in the conterminous United States: Utilizing the special report on emission scenarios at ecoregional scales. Glob Environ Change 22: 896-914. doi: 10.1016/j.gloenvcha.2012.03.008
|
| [43] | Sleeter BM, Wilson TS, Soulard CE, et al. (2010) Estimation of late twentieth century land-cover change in California. Environ Monit Assess 173: 251-266. |
| [44] |
Sleeter BM, Sohl TL, Loveland TR, et al. (2013) Land-cover change in the conterminous United States from 1973 to 2000. Glob Environ Change 23: 733-748. doi: 10.1016/j.gloenvcha.2013.03.006
|
| [45] | Daniel CJ, Frid L. ST-Sim - State-and-Transition Simulation Model. Apex Resource Management Solutions. Available from: http://www.apexrms.com/stsm |
| [46] | Kerns BK, Shlisky AJ, Daniel CJ (2012) Proceedings of the First Landscape State-and-Transition Simulation Modeling Conference, June 14-16, 2011, Portland, Oregon. |
| [47] | Soil Survey Staff, Natural Resources Conservation Service Web Soil Survey (SSURGO). Available from: http://www.nrcs.usda.gov/wps/portal/nrcs/detail/soils/survey/geo/?cid=nrcs142p2_053627. |
| [48] | Sleeter RR, Gould M (2007) Geographic Information System Software to Remodel Population Data Using Dasymetric Mapping Methods. U.S. Geological Survey Techniques and Methods 11-C2. Reston, Virginia. Available from: http://pubs.usgs.gov/tm/tm11c2/. |
| [49] | Sleeter RR, Acevedo C, Soulard CE, et al. (2015) Methods used to parameterize the spatially-explicit components of a state-and-transition simulation model. AIMS Environ Sci [submitted]. |
| [50] | Spencer WD, Beier P, Penrod K, et al. (2010) California Essential Habitat Connectivity Project: A Strategy for Conserving a Connected California. Prepared for California Department of Transportation, California Department of Fish and Game, and Federal Highways Administration. Available from: https://www.wildlife.ca.gov/Conservation/Planning/Connectivity/CEHC. |
| [51] | Norum K (2011) Agricultural Water Use in California: A 2011 Update. The Center for Irrigation Technology, California State University, Fresno. |
| [52] |
Theobald DM (2003) Targeting Conservation Action through Assessment of Protection and Exurban Threats. Conserv Biol 17: 1624-1637. doi: 10.1111/j.1523-1739.2003.00250.x
|
| [53] |
Wade AA, Theobald DM, Laituri MJ (2011) A multi-scale assessment of local and contextual threats to existing and potential U.S. protected areas. Landsc Urban Plan 101: 215-227. doi: 10.1016/j.landurbplan.2011.02.027
|
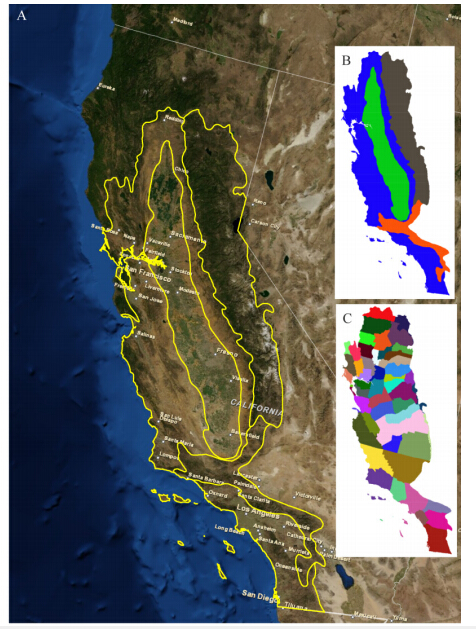
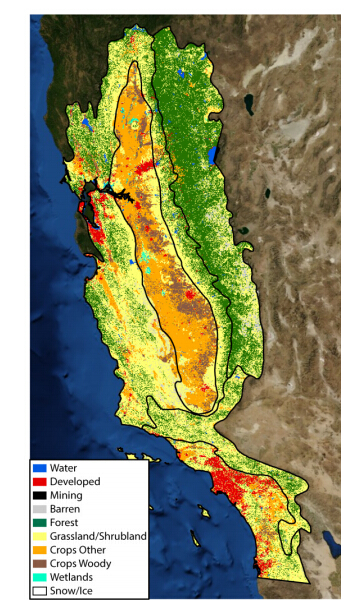
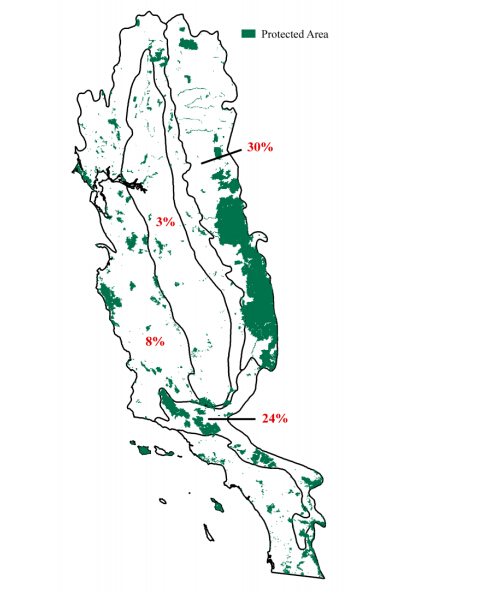
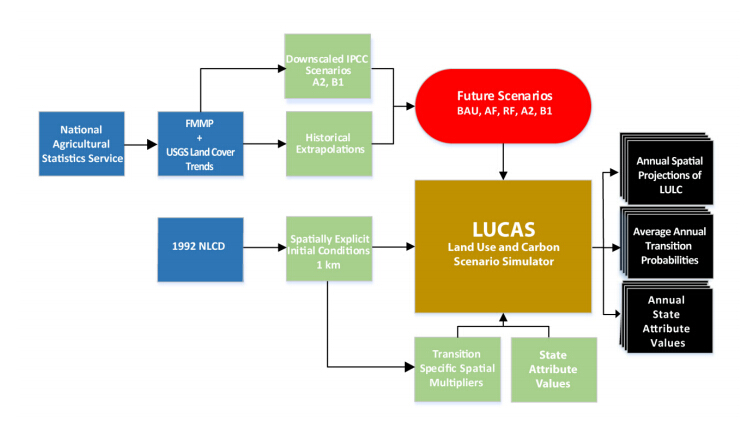
Figures(9) / Tables(3)

Tamara S. Wilson, Benjamin M. Sleeter, Jason Sherba, Dick Cameron. Land-use impacts on water resources and protected areas: applications of state-and-transition simulation modeling of future scenarios[J]. AIMS Environmental Science, 2015, 2(2): 282-301. doi: 10.3934/environsci.2015.2.282







 DownLoad:
DownLoad: