Citation: Gerald Pawlish, Kyle Spivack, Adam Gabriel, Zuyi Huang, Noelle Comolli. Chemotherapeutic loading via tailoring of drug-carrier interactions in poly (sialic acid) micelles[J]. AIMS Bioengineering, 2018, 5(2): 106-132. doi: 10.3934/bioeng.2018.2.106

| [1] | Caliceti P, Salmaso S (2013) Stealth properties to improve therapeutic efficacy of drug nanocarriers. J Drug Deli 2013: 1–19. |
| [2] |
Jayant S, Khandare JJ, Wang Y, et al. (2007) Targeted sialic acid-doxorubicin prodrugs for intracellular delivery and cancer treatment. Pharm Res 24: 2120–2130. doi: 10.1007/s11095-007-9406-1

|
| [3] |
Li Y, Yang L (2015) Driving forces for drug loading in drug carriers. J Microencapsu 32: 255–272. doi: 10.3109/02652048.2015.1010459
|
| [4] |
Wang XH, Tian Q, Wang W, et al. (2012) In vitro evaluation of polymeric micelles based on hydrophobically-modified sulfated chitosan as a carrier of doxorubicin. J Mater Sci-Mater M 23: 1663–1674. doi: 10.1007/s10856-012-4627-1
|
| [5] |
Li JF, Yang HY, Zhang YJ, et al. (2015) Choline Derivate-Modified Doxorubicin Loaded Micelle for Glioma Therapy. Acs Appl Mater Inter 7: 21589–21601. doi: 10.1021/acsami.5b07045
|
| [6] |
Su Y, Hu Y, Du Y, et al. (2015) Redox-responsive polymer drug conjugates based on doxorubicin and chitosan oligosaccharide-g-stearic acid for cancer therapy. Mol Pharm 12: 1193–1202. doi: 10.1021/mp500710x
|
| [7] |
Gou P, Liu W, Mao W, et al. (2013) Self-assembling doxorubicin prodrug forming nanoparticles for cancer chemotherapy: Synthesis and anticancer study in vitro and in vivo. J Mater Chem B 1: 284–292. doi: 10.1039/C2TB00004K
|
| [8] |
Sun Y, Zou W, Bian SQ, et al. (2013) Bioreducible PAA-g-PEG graft micelles with high doxorubicin loading for targeted antitumor effect against mouse breast carcinoma. Biomaterials 34: 6818–6828. doi: 10.1016/j.biomaterials.2013.05.032
|
| [9] |
Jain RK, Stylianopoulos T (2010) Delivering nanomedicine to solid tumors. Nat Rev Clin Oncol 7: 653–664. doi: 10.1038/nrclinonc.2010.139
|
| [10] |
Du YZ, Weng Q, Yuan H, et al. (2010) Synthesis and antitumor activity of stearate-g-dextran micelles for intracellular doxorubicin delivery. Acs Nano 4: 6894–6902. doi: 10.1021/nn100927t
|
| [11] |
Wang YC, Wang F, Sun TM, et al. (2011) Redox-responsive nanoparticles from the single disulfide bond-bridged block copolymer as drug carriers for overcoming multidrug resistance in cancer cells. Bioconjugate Chem 22: 1939–1945. doi: 10.1021/bc200139n
|
| [12] |
Zhang AP, Zhang Z, Shi FH, et al. (2013) Redox-sensitive shell-crosslinked polypeptide-block-polysaccharide micelles for efficient intracellular anticancer drug delivery. Macromol Biosci 13: 1249–1258. doi: 10.1002/mabi.201300175
|
| [13] |
Shuai XT, Ai H, Nasongkla N, et al. (2004) Micellar carriers based on block copolymers of poly(e-caprolactone) and poly(ethylene glycol) for doxorubicin delivery. J Control Release 98: 415–426. doi: 10.1016/j.jconrel.2004.06.003
|
| [14] |
Song HT, Hoang NH, Yun JM, et al. (2016) Development of a new tri-block copolymer with a functional end and its feasibility for treatment of metastatic breast cancer. Colloids Surface B 144: 73–80. doi: 10.1016/j.colsurfb.2016.04.002
|
| [15] |
Zhao XY, Poon Z, Engler AC, et al. (2012) Enhanced stability of polymeric micelles based on postfunctionalized poly(ethylene glycol)-b-poly(gamma-propargyl L-glutamate): The substituent effect. Biomacromolecules 13: 1315–1322. doi: 10.1021/bm201873u
|
| [16] |
Hamaguchi T, Matsumura Y, Suzuki M, et al. (2005) NK105, a paclitaxel-incorporating micellar nanoparticle formulation, can extend in vivo antitumour activity and reduce the neurotoxicity of paclitaxel. Brit J Cancer 92: 1240–1246. doi: 10.1038/sj.bjc.6602479
|
| [17] |
Shi Y, van Steenbergen MJ, Teunissen EA, et al. (2013) Pi-Pi stacking increases the stability and loading capacity of thermosensitive polymeric micelles for chemotherapeutic drugs. Biomacromolecules 14: 1826–1837. doi: 10.1021/bm400234c
|
| [18] |
Carstens MG, de Jong P, van Nostrum CF, et al. (2008) The effect of core composition in biodegradable oligomeric micelles as taxane formulations. Eur J Pharm Biopharm 68: 596–606. doi: 10.1016/j.ejpb.2007.08.014
|
| [19] |
Yokoyama M, Fukushima S, Uehara R, et al. (1998) Characterization of physical entrapment and chemical conjugation of adriamycin in polymeric micelles and their design for in vivo delivery to a solid tumor. J Control Release 50: 79–92. doi: 10.1016/S0168-3659(97)00115-6
|
| [20] |
Kataoka K, Matsumoto T, Yokoyama M, et al. (2000) Doxorubicin-loaded poly(ethylene glycol)-poly(beta-benzyl-l-aspartate) copolymer micelles: Their pharmaceutical characteristics and biological significance. J Control Release 64: 143–153. doi: 10.1016/S0168-3659(99)00133-9
|
| [21] |
Hofman JW, Carstens MG, van Zeeland F, et al. (2008) Photocytotoxicity of mTHPC (temoporfin) loaded polymeric micelles mediated by lipase catalyzed degradation. Pharm Res 25: 2065–2073. doi: 10.1007/s11095-008-9590-7
|
| [22] |
Mikhail AS, Allen C (2010) Poly(ethylene glycol)-b-poly(epsilon-caprolactone) micelles containing chemically conjugated and physically entrapped docetaxel: Synthesis, characterization, and the influence of the drug on micelle morphology. Biomacromolecules 11: 1273–1280. doi: 10.1021/bm100073s
|
| [23] |
Wang C, Chen B, Zou M, et al. (2014) Cyclic RGD-modified chitosan/graphene oxide polymers for drug delivery and cellular imaging. Colloid Surface B 122: 332–340. doi: 10.1016/j.colsurfb.2014.07.018
|
| [24] |
Yan JL, Ye ZY, Chen M, et al. (2011) Fine tuning micellar core-forming block of poly(ethylene glycol)-block-poly(epsilon-caprolactone) amphiphilic copolymers based on chemical modification for the solubilization and delivery of doxorubicin. Biomacromolecules 12: 2562–2572. doi: 10.1021/bm200375x
|
| [25] |
Bader RA, Silvers AL, Zhang N (2011) Polysialic acid-based micelles for encapsulation of hydrophobic drugs. Biomacromolecules 12: 314–320. doi: 10.1021/bm1008603
|
| [26] |
Veronese FM, Pasut G (2005) PEGylation, successful approach to drug delivery. Drug Discov Today 10: 1451–1458. doi: 10.1016/S1359-6446(05)03575-0
|
| [27] |
Deepagan VG, Thambi T, Ko H, et al. (2013) Amphiphilic polysialic acid derivatives: Synthesis, characterization, and in-vitro cytotoxicity. J Nanosci Nanotechno 13: 7312–7318. doi: 10.1166/jnn.2013.8089
|
| [28] |
Gregoriadis G, McCormack B, Wang Z, et al. (1993) Polysialic acids-potential in drug delivery. Febs Lett 315: 271–276. doi: 10.1016/0014-5793(93)81177-2
|
| [29] |
Konradi R, Acikgoz C, Textor M (2012) Polyoxazolines for nonfouling surface coatings-a direct comparison to the gold standard PEG. Macromol Rapid Comm 33: 1663–1676. doi: 10.1002/marc.201200422
|
| [30] |
Bondioli L, Ruozi B, Belletti D, et al. (2011) Sialic acid as a potential approach for the protection and targeting of nanocarriers. Expert Opin Drug Del 8: 921–937. doi: 10.1517/17425247.2011.577061
|
| [31] | Semple SC, Harasym TO, Clow KA, et al. (2005) Immunogenicity and rapid blood clearance of liposomes containing polyethylene glycol-lipid conjugates and nucleic acid. J Pharmacol Exp Ther 312: 1020–1026. |
| [32] |
Amoozgar Z, Yeo Y (2012) Recent advances in stealth coating of nanoparticle drug delivery systems. Wires Nanomed Nanobio 4: 219–233. doi: 10.1002/wnan.1157
|
| [33] |
Zhang N, Wardwell PR, Bader RA (2013) Polysaccharide-based micelles for drug delivery. Pharmaceutics 5: 329–352. doi: 10.3390/pharmaceutics5020329
|
| [34] |
Wilson DR, Zhang N, Silvers AL, et al. (2014) Synthesis and evaluation of cyclosporine A-loaded polysialic acid-polycaprolactone micelles for rheumatoid arthritis. Eur J Pharm Sci 51: 146–156. doi: 10.1016/j.ejps.2013.09.013
|
| [35] |
Bondioli L, Costantino L, Ballestrazzi A, et al. (2010) PLGA nanoparticles surface decorated with the sialic acid, N-acetylneuraminic acid. Biomaterials 31: 3395–3403. doi: 10.1016/j.biomaterials.2010.01.049
|
| [36] |
Deninno MP (1991) The synthesis and glycosidation of n-acetylneuraminic acid. Synthesis 1991: 583–593. doi: 10.1055/s-1991-26522
|
| [37] |
Rutishauser U (1998) Polysialic acid at the cell surface: Biophysics in service of cell interactions and tissue plasticity. J Cell Biochem 70: 304–312. doi: 10.1002/(SICI)1097-4644(19980901)70:3<304::AID-JCB3>3.0.CO;2-R
|
| [38] |
Vodovozova EL, Moiseeva EV, Grechko GK, et al. (2000) Antitumour activity of cytotoxic liposomes equipped with selectin ligand SiaLe(X), in a mouse mammary adenocarcinoma model. Eur J Cancer 36: 942–949. doi: 10.1016/S0959-8049(00)00029-0
|
| [39] |
Hirai M, Minematsu H, Hiramatsu Y, et al. (2010) Novel and simple loading procedure of cisplatin into liposomes and targeting tumor endothelial cells. Int J Pharm 391: 274–283. doi: 10.1016/j.ijpharm.2010.02.030
|
| [40] |
Zheng JS, Zheng SY, Zhang YB, et al. (2011) Sialic acid surface decoration enhances cellular uptake and apoptosis-inducing activity of selenium nanoparticles. Colloid Surface B 83: 183–187. doi: 10.1016/j.colsurfb.2010.11.023
|
| [41] |
Greco F, Arif I, Botting R, et al. (2013) Polysialic acid as a drug carrier: evaluation of a new polysialic acid-epirubicin conjugate and its comparison against established drug carriers. Polym Chem-UK 4: 1600–1609. doi: 10.1039/C2PY20876H
|
| [42] |
Zeisig R, Stahn R, Wenzel K, et al. (2004) Effect of sialyl Lewis X-glycoliposomes on the inhibition of E-selectin-mediated tumour cell adhesion in vitro. Biochim Biophys Acta 1660: 31–40. doi: 10.1016/j.bbamem.2003.10.014
|
| [43] |
Gajbhiye V, Ganesh N, Barve J, et al. (2013) Synthesis, characterization and targeting potential of zidovudine loaded sialic acid conjugated-mannosylated poly(propyleneimine) dendrimers. Eur J Pharm Sci 48: 668–679. doi: 10.1016/j.ejps.2012.12.027
|
| [44] |
Zhang WX, Dong DQ, Li P, et al. (2016) Novel pH-sensitive polysialic acid based polymeric micelles for triggered intracellular release of hydrophobic drug. Carbohyd Polym 139: 75–81. doi: 10.1016/j.carbpol.2015.12.041
|
| [45] |
Ray GB, Chakraborty I, Moulik SP (2006) Pyrene absorption can be a convenient method for probing critical micellar concentration (cmc) and indexing micellar polarity. J Colloid Interf Sci 294: 248–254. doi: 10.1016/j.jcis.2005.07.006
|
| [46] |
Hans M, Shimoni K, Danino D, et al. (2005) Synthesis and characterization of mPEG-PLA prodrug micelles. Biomacromolecules 6: 2708–2717. doi: 10.1021/bm050188k
|
| [47] |
Liang HC, Chang WH, Liang HF, et al. (2004) Crosslinking structures of gelatin hydrogels crosslinked with genipin or a water-soluble carbodiimide. J Appl Polym Sci 91: 4017–4026. doi: 10.1002/app.13563
|
| [48] |
Koo AN, Min KH, Lee HJ, et al. (2012) Tumor accumulation and antitumor efficacy of docetaxel-loaded core-shell-corona micelles with shell-specific redox-responsive cross-links. Biomaterials 33: 1489–1499. doi: 10.1016/j.biomaterials.2011.11.013
|
| [49] |
Lee H, Ahn CH, Park TG (2009) Poly lactic-co-(glycolic acid)-grafted hyaluronic acid copolymer micelle nanoparticles for target-specific delivery of doxorubicin. Macromol Biosci 9: 336–342. doi: 10.1002/mabi.200800229
|
| [50] |
Sutton D, Wang S, Nasongkia N, et al. (2007) Doxorubicin and beta-lapachone release and interaction with micellar core materials: experiment and modeling. Exp Biol Med 232: 1090–1099. doi: 10.3181/0702-RM-31
|
| [51] |
Donaldson O, Huang Z, Comolli N (2013) An integrated experimental and modeling approach to propose biotinylated PLGA microparticles as versatile targeting vehicles for drug delivery. Prog Biomat 2: 1–10. doi: 10.1186/2194-0517-2-1
|
| [52] |
Zhai S, Ma Y, Chen Y, et al. (2014) Synthesis of an amphiphilic block copolymer containing zwitterionic sulfobetaine as a novel pH-sensitive drug carrier. Polym Chem 5: 1285–1297. doi: 10.1039/C3PY01325A
|
| [53] |
Deshmukh AS, Chauhan PN, Malleshappa NN, et al. (2017) Polymeric micelles: Basic research to clinical practice. Int J Pharm 532: 249–268. doi: 10.1016/j.ijpharm.2017.09.005
|
| [54] |
Aftab S, Shah A, Nadhman A, et al. (2018) Nanomedicine: An effective tool in cancer therapy. Int J Pharm 540: 132–149. doi: 10.1016/j.ijpharm.2018.02.007
|
 bioeng-05-02-106-s01.pdf bioeng-05-02-106-s01.pdf |
 |
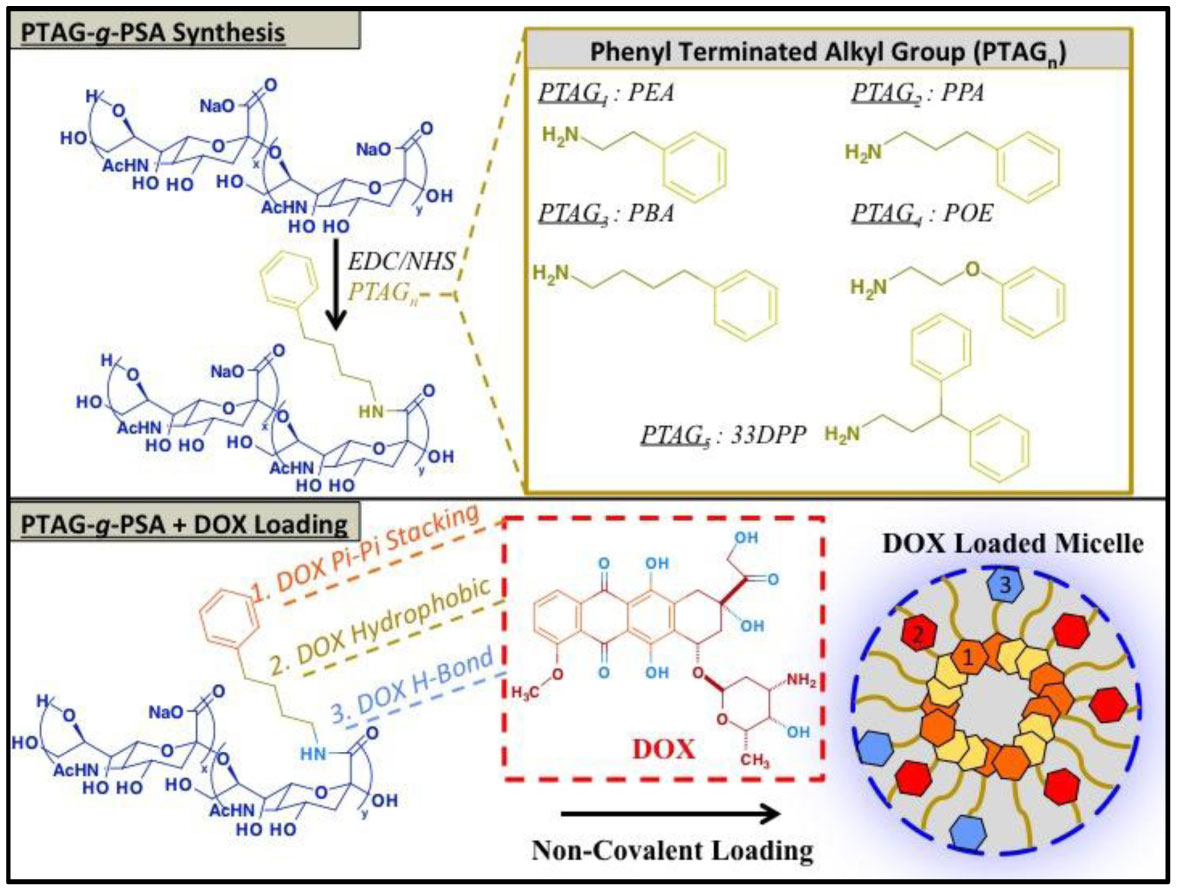
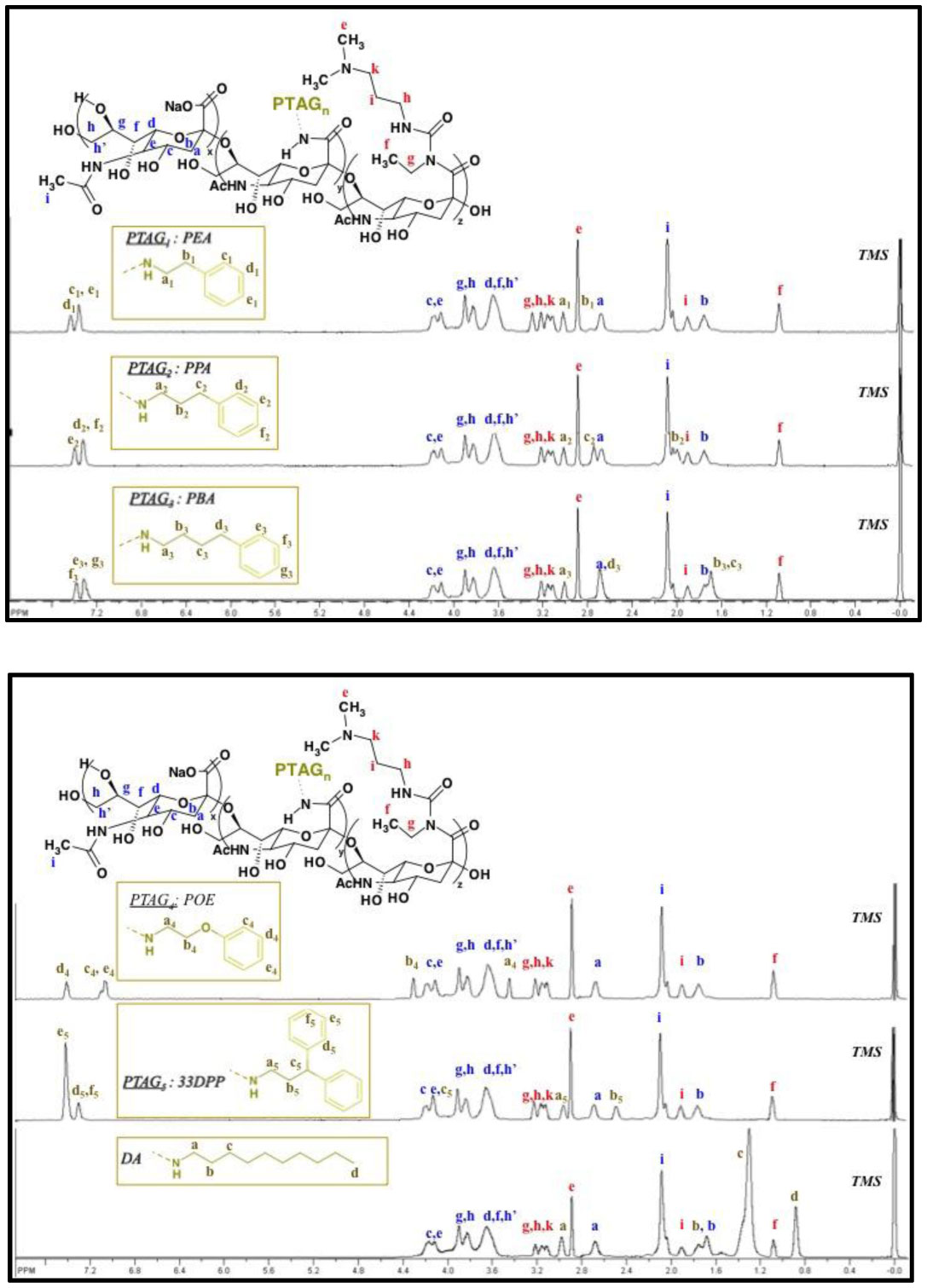

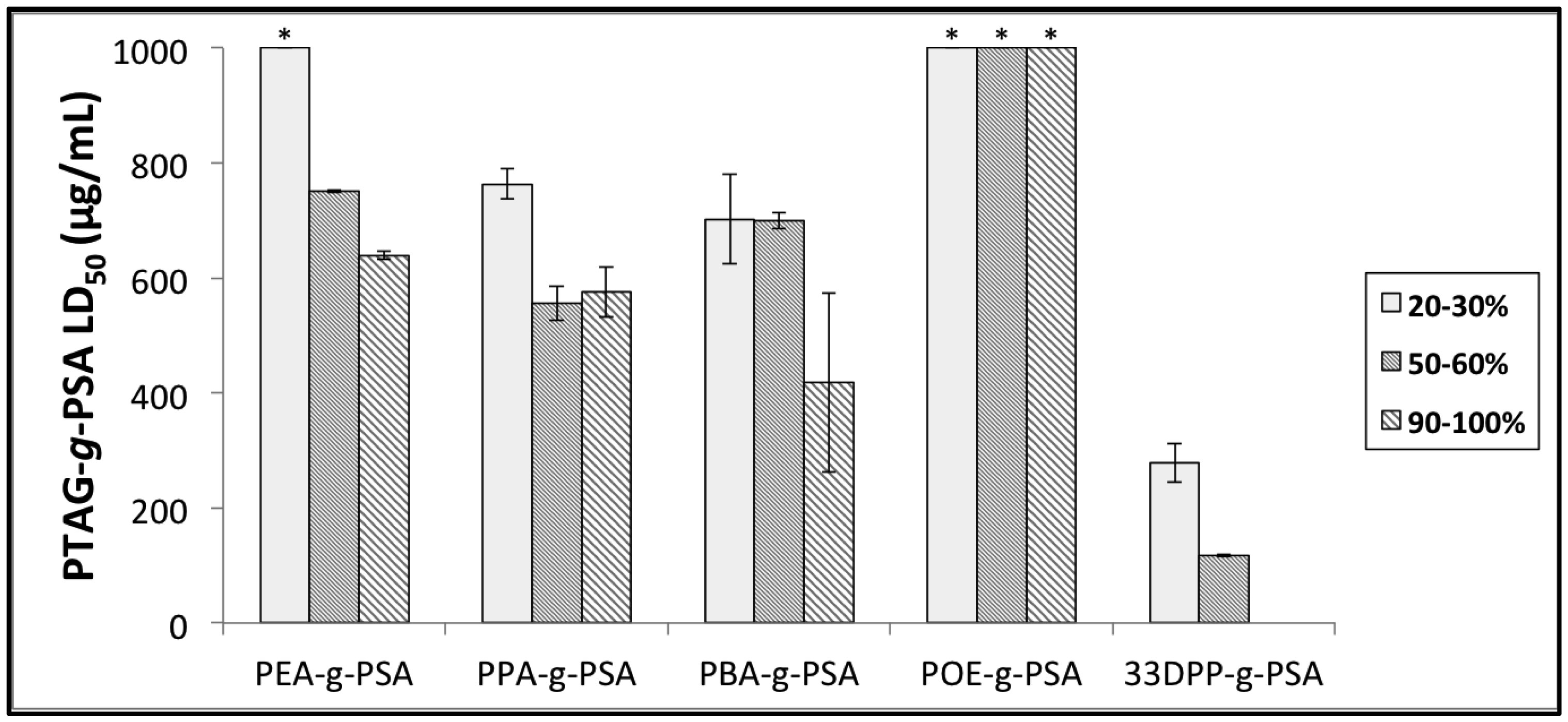

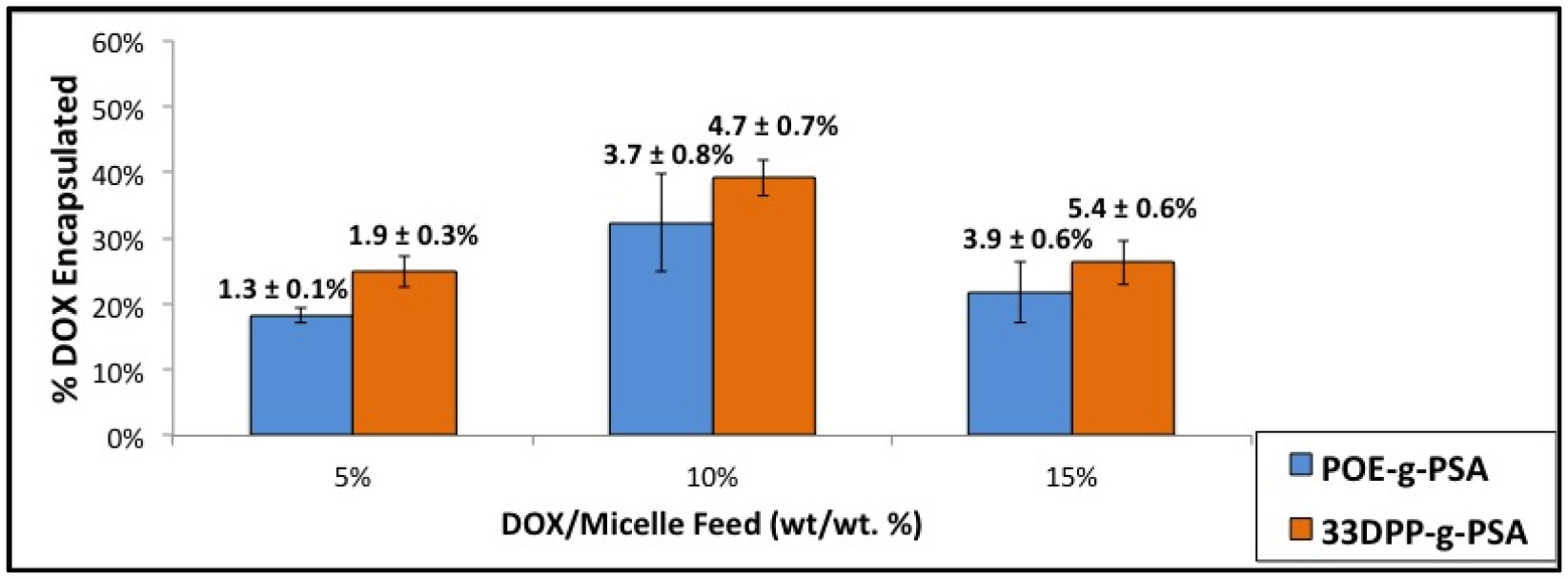
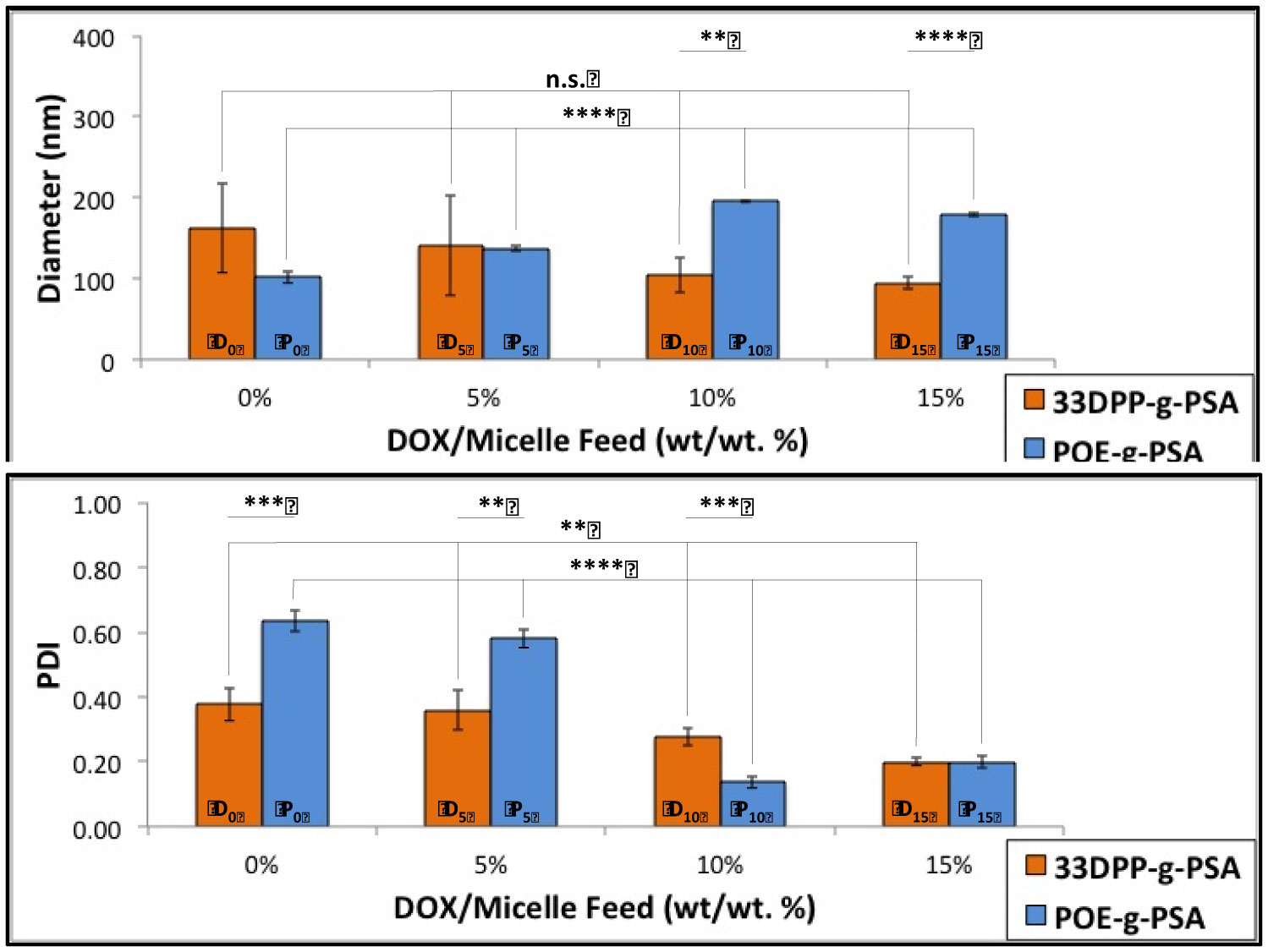
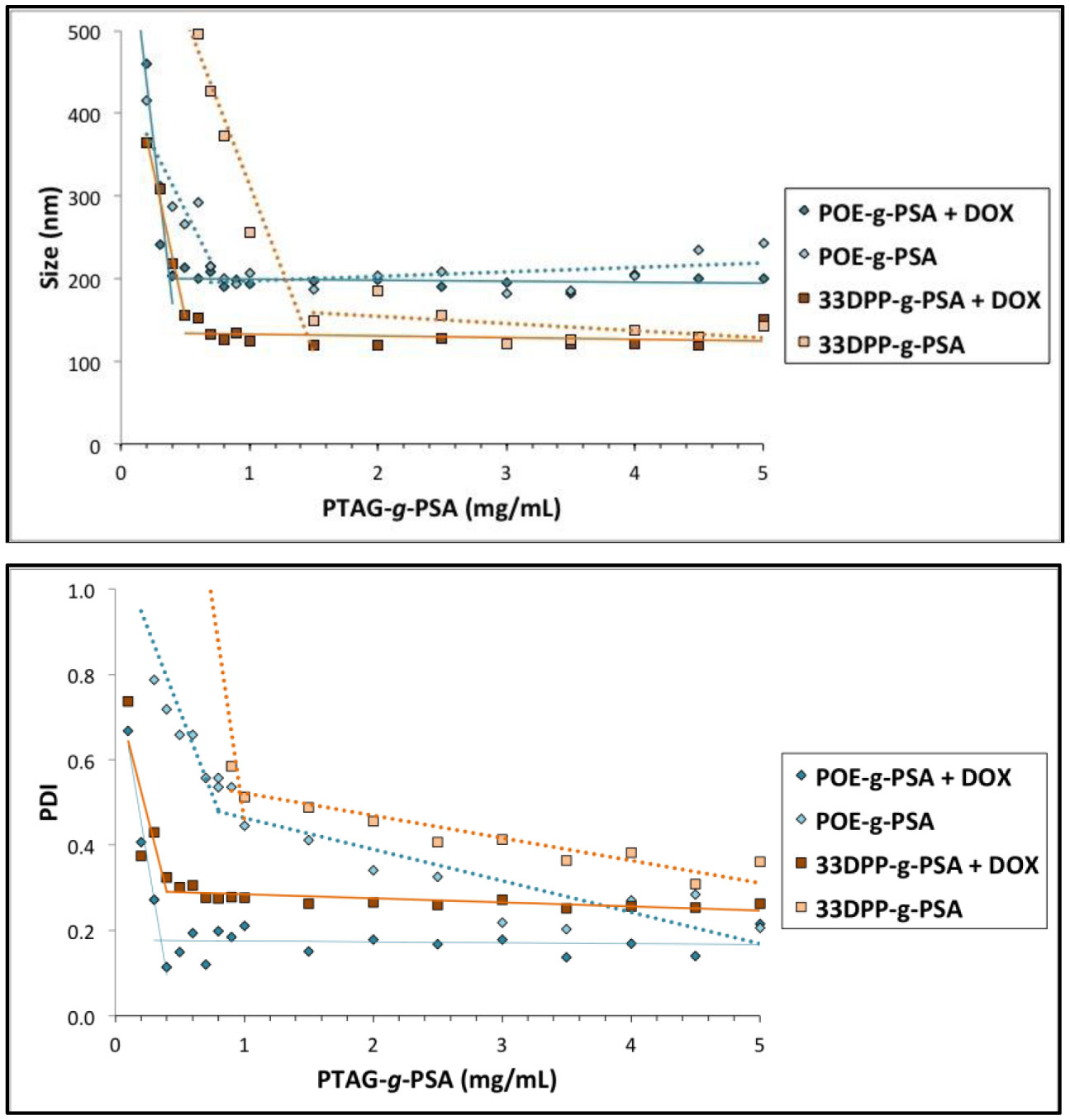
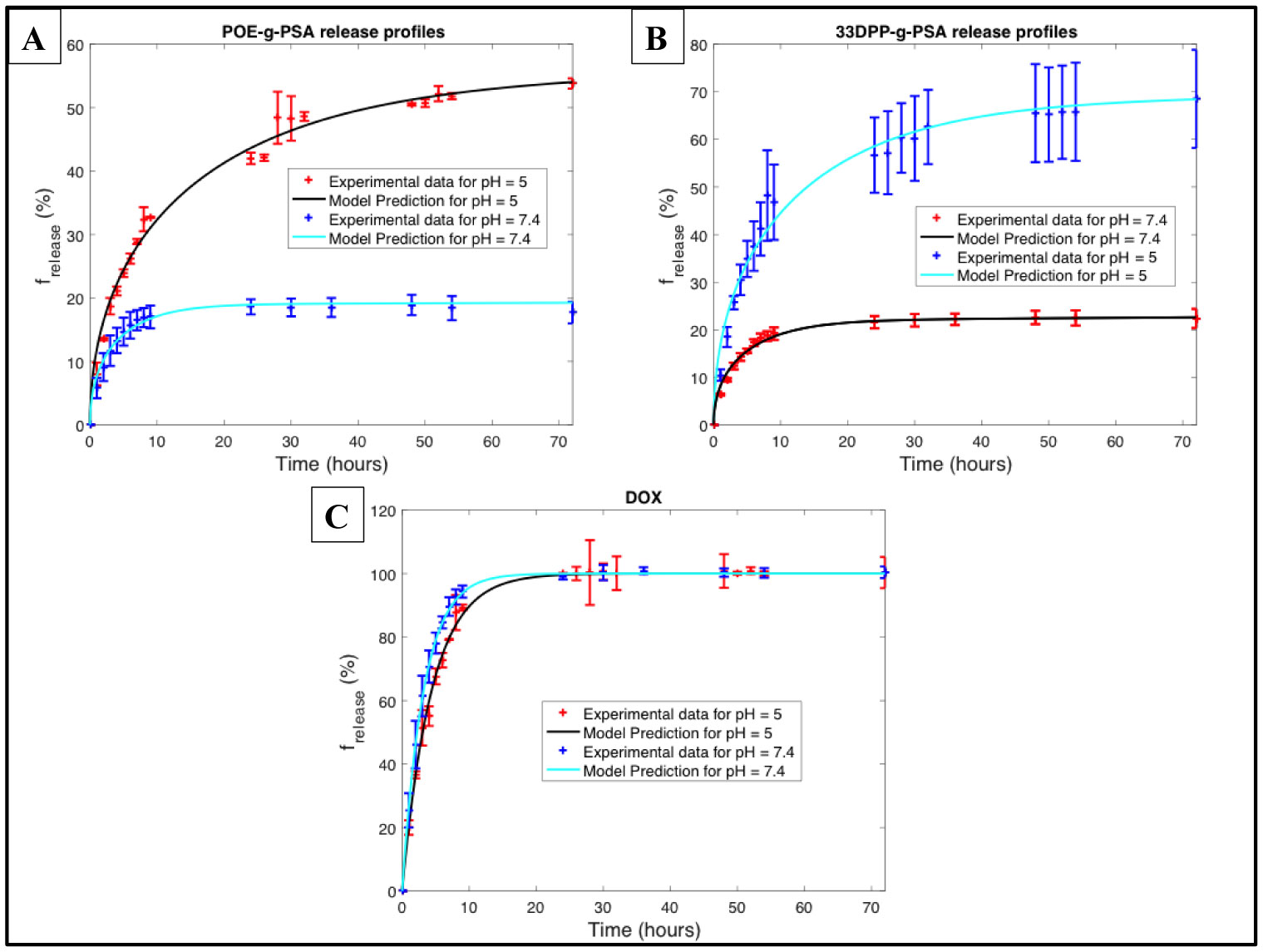
Figures(9) / Tables(3)

Gerald Pawlish, Kyle Spivack, Adam Gabriel, Zuyi Huang, Noelle Comolli. Chemotherapeutic loading via tailoring of drug-carrier interactions in poly (sialic acid) micelles[J]. AIMS Bioengineering, 2018, 5(2): 106-132. doi: 10.3934/bioeng.2018.2.106






 DownLoad:
DownLoad: