Citation: Edwige Arnold, Iness Hammami, Jingkui Chen, Sachin Gupte, Yves Durocher, Mario Jolicoeur. Overexpression of G6PDH does not affect the behavior of HEK-293 clones stably expressing interferon-α2b[J]. AIMS Bioengineering, 2016, 3(3): 319-336. doi: 10.3934/bioeng.2016.3.319

| [1] | Kamen A, Henry O (2004) Development and optimization of an adenovirus production process. J Gene Med 6: S184–S192. |
| [2] | Durocher Y, Pham PL, St-Laurent G, et al. (2007) Scalable serum-free production of recombinant adeno-associated virus type 2 by transfection of 293 suspension cells. J Virol Methods 144: 32–40. |
| [3] | Baldi L, Muller N, Picasso S, et al. (2005) Transient gene expression in suspension HEK-293 cells: application to large-scale protein production. Biotechnol Progr 21: 148–153. |
| [4] | Henry O, Durocher Y (2011) Enhanced glycoprotein production in HEK-293 cells expressing pyruvate carboxylase. Metab Eng 13: 499–507. |
| [5] | Elias CB, Carpentier E, Durocher Y, et al. (2003) Improving glucose and glutamine metabolism of human HEK 293 and Trichoplusia ni insect cells engineered to express a cytosolic pyruvate carboxylase enzyme. Biotechnol Progr 19: 90–97. |
| [6] | Loignon M, Perret S, Kelly J, et al. (2008) Stable high volumetric production of glycosylated human recombinant IFNalpha2b in HEK293 cells. BMC Biotechnol 8: 65. |
| [7] | Klein T, Niklas J, Heinzle E (2015) Engineering the supply chain for protein production/secretion in yeasts and mammalian cells. J Ind Microbiol Biot 42: 453–464. |
| [8] | Dietmair S, Nielsen LK, Timmins NE (2011) Engineering a mammalian super producer. J Chem Tech Biot 86: 905–914. |
| [9] | Costa AR, Rodrigues ME, Henriques M, et al. (2010) Guidelines to cell engineering for monoclonal antibody production. Eur J Pharm Biopharm 74: 127–138. |
| [10] | Vallée C, Durocher Y, Henry O (2014) Exploiting the metabolism of PYC expressing HEK293 cells in fed-bach cultures. J Biotechnol 169: 63–70. |
| [11] | Seth G, Hossler P, Yee JC, et al. (2006) Engineering cells for cell culture bioprocessing--physiological fundamentals. Adv Biochem Eng Biotechnol 101: 119–164. |
| [12] | Templeton N, Dean J, Reddy P, et al. (2013) Peak antibody production is associated with increased oxidative metabolism in an industrially relevant fed-batch CHO cell culture. Biotechnol Bioeng 110: 2013–2024. |
| [13] | Ahn WS, Antoniewicz MR (2011) Metabolic flux analysis of CHO cells at growth and non-growth phases using isotopic tracers and mass spectrometry. Metab Eng 12: 598–609. |
| [14] | Sengupta N, Rose ST, Morgan JA (2011) Metabolic Flux Analysis of CHO Cell Metabolism in the Late Non-Growth Phase. Biotechnol Bioeng 108: 82–92. |
| [15] | Dean J, Reddy P (2013) Metabolic analysis of antibody producing CHO cells in fed-batch production. Biotechnol Bioeng 110: 1735–1747. |
| [16] | Sheikholeslami Z, Jolicoeur M, Henry O (2013) The impact of the timing of induction on the metabolism and productivity of CHO cells in culture. Biochem Eng J 79: 162–171. |
| [17] | Fang H, Xie X, Xu Q, et al. (2013) Enhancement of cytidine production by coexpression of gnd, zwf, and prs genes in recombinant Escherichia coli CYT15 Biotechnol Lett 35: 245–251. |
| [18] | Shi F, Li K, Huan X, et al. (2013) Expression of NAD(H) kinase and glucose-6- phosphate dehydrogenase improve NADPH supply and L-isoleucine biosynthesis in corynebacterium glutamicum ssp. lactofermentum. Appl Microbiol Biot 171: 504–521. |
| [19] | Flores S, Anda-Herrera RD, Gosset G, et al. (2004) Growth-rate recovery of Escherichia coli cultures carrying a multicopy plasmid, by engineering of the pentose- phosphate pathway. Biotechnol Bioeng 87: 485–494. |
| [20] | Kwon D-H, Kim MD, Lee TH, et al. (2006) Elevation of glucose 6-phosphate dehydrogenase activity increases xylitol production in recombinant Saccharomyces cerevisiae. J Mol Cat B: Enz 43: 86–89. |
| [21] | Wang XT, Engel PC (2009) An optimised system for refolding of human glucose 6-phosphate dehydrogenase. BMC Biotechnol 9–19. |
| [22] | Tian W-N, Braunstein LD, Pang J, et al. (1998) Importance of glucose-6-phosphate dehydrogenase activity for cell growth. J Biol Chem 273: 10609–10617. |
| [23] | Duan YX, Chen T, Chen X, et al. (2010) Overexpression of glucose-6-phosphate dehydrogenase enhances riboflavin production in Bacillus subtilis. Appl Microbiol Biot 85: 1907–1914. |
| [24] | Stanton RC (2012) Glucose-6-phosphate dehydrogenase, NADPH, and cell survival. IUBMB Life 64: 363–369. |
| [25] | Pandolfi PP, Sonati F, Rivi R, et al. (1995) Targeted disruption of the housekeeping gene encoding glucose 6-phosphate dehydrogenase oxidative stress. EMBO J 14: 5209–5215. |
| [26] | Zhang Z, Yang Z, Zhu B, et al. (2012) Increasing glucose 6-phosphate dehydrogenase activity restores redox balance in vascular endothelial cells exposed to high glucose. PLoS ONE 7: e49128. |
| [27] | Ursini MV, Parrella A, Rosa G, et al. (1997) Enhanced expression of glucose-6-phosphate dehydrogenase in human cells sustaining oxidative stress. Biochem J 323: 801–806. |
| [28] | Tian WN, Braunstein LD, Apse K, et al. (1999) Importance of glucose-6-phosphate dehydrogenase activity in cell death. Am J Physiol 276: C1121–1131. |
| [29] | Ghorbaniaghdam A, Henry O, Jolicoeur M (2014) An in-silico study of the regulation of CHO cells glycolysis. J Theor Biol 357: 112–122. |
| [30] | Gupte RS, Ata H, Rawat D, et al. (2011) Glucose-6-phosphate dehydrogenase is a regulator of vascular smooth muscle contraction. Antioxydants Red Signal 14: 543–558. |
| [31] | Sacchetti A, Subramaniam V, Jovin TM, et al. (2002) Oligomerization of DsRed is required for the generation of a functional red fluorescent chromophore. FEBS Lett 525: 13–19. |
| [32] | Tom R, Bisson L, Durocher Y (2007) Expression Systems: Methods Express, Scion Publishing Ltd. |
| [33] | Ghorbaniaghdam A, Chen J, Henry O, et al. (2014) Analyzing clonal variation of monoclonal antibody-producing CHO cell lines using an in silico metabolomic platform. PLoS ONE 9. |
| [34] | Boucher C, St-Laurent G, Loignon M, et al. (2008) The bioactivity and receptor affinity of recombinant tagged EGF designed for tissue engineering applications is defined by the nature and position of the tags. Tissue Eng Part A 14: 2069–2077. |
| [35] | Cotlet M, Hofkens J, Habuchi S, et al. (2001) Identification of different emitting species in the red fluorescent protein DsRed by means of ensemble and single-molecule spectroscopy. Proc Natl Acad Sci USA 98: 14398–14403. |
| [36] | Baird GS, Zacharias DA, Tsien RY (2000) Biochemistry, mutagenesis, and oligomerization of DsRed, a red fluorescent protein from coral. Proc Natl Acad Sci USA 97: 11984–11989. |
| [37] | Nicolas C, Kiefer P, Letisse F, et al. (2007) Response of the central metabolism of Escherichia coli to modified expression of the gene encoding the glucose-6-phosphate dehydrogenase. FEBS Lett 581: 3771–3776. |
| [38] | Cruz HJ, Freitas CM, Alves PM, et al. (2000) Effect of ammonia and lactate on growth, metabolism, and productivity of BHK cells. Enzyme Microb Tech 27: 3–52. |
| [39] | Dietmair S, Hodson MP, Quek LE, et al. (2012) A Multi- Omics Analysis of Recombinant Protein Production in Hek293 Cells. PLoS ONE 7: 8, e43394. |
| [40] | Templeton N, Dean J, Reddy P, et al. (2013) Peak Antibody Production is Associated With Increased Oxidative Metabolism in an Industrially Relevant Fed- Batch CHO Cell Culture. Biotechnol Bioeng 110: 2013–2024. |
| [41] | Pollak N, Niere M, Ziegler M (2007) NAD kinase levels control the NADPH concentration in human cells. J Biol Chem 282: 33562–33571. |
| [42] | Hardie DG (2004) The AMP-activated protein kinase pathway - new players upstream and downstream. J Cell Sci 117: 5479–5487. |
| [43] | Irani N, Wirth M, Heuvel JVD, et al. (1999) Improvement of the primary metabolism of cell cultures by introducing a new cytoplasmic pyruvate carboxylase reaction. Biotechnol Bioeng 66: 238–246. |
| [44] | Chong WPK, Thng SH, Hiu AP, et al. (2012) LC-MS-Based Metabolic Characterization of High Monoclonal Antibody-Producing Chinese Hamster Ovary Cells. Biotechnol Bioeng 109: 3103–3111. |
| [45] | Ying W (2008) NAD+/NADH and NADP+/NADPH in cellular functions and cell death: regulation and biological consequences. Antioxydants Redox Signal 10: 179–206. |
| [46] | Wilkens CA, Gerdtzen ZP (2015) Comparative Metabolic Analysis of CHO Cell Clones Obtained through Cell Engineering, for IgG Productivity, Growth and Cell Longevity. PLoS ONE 10: e0119053. |
| [47] | Wilkens CA, Altamirano C, Gerdtzen ZP (2011) Comparative metabolic analysis of lactate for CHO cells in glucose and galactose. Biotechnol Bioproc Eng 16: 714–724. |
| [48] | Lane AN, Fan TW-M (2015) Regulation of mammalian nucleotide metabolism and biosynthesis. Nucleic Acids Res 43: 2466–2485. |
| [49] | Luo J, Vijayasankaran N, Autsen J, et al. (2012) Comparative metabolite analysis to understand lactate metabolism shift in Chinese hamster ovary cell culture process. Biotechnol Bioeng 109: 146–156. |
| [50] | Sellick CA, Croxford AS, Maqsood AR, et al. (2011) Metabolite profiling of Recombinant CHO Cells: Designing Tailored Feeding Regimes That Enhance Recombinant Antibody Production. Biotechnol Bioeng 108: 3025–3031. |
| [51] | Carinhas N, Duarte TM, Barreiro LC, et al. (2013) Metabolic Signatures of GS-CHO Cell Clones Associated With Butyrate Treatment and Culture Phase Transition. Biotechnol Bioeng 110: 3244–3257. |
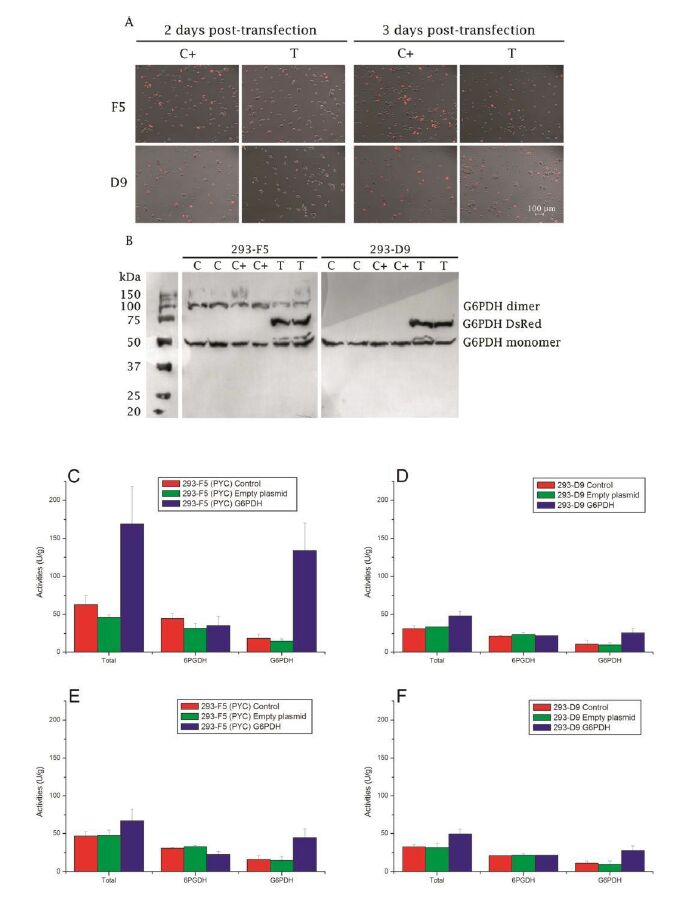
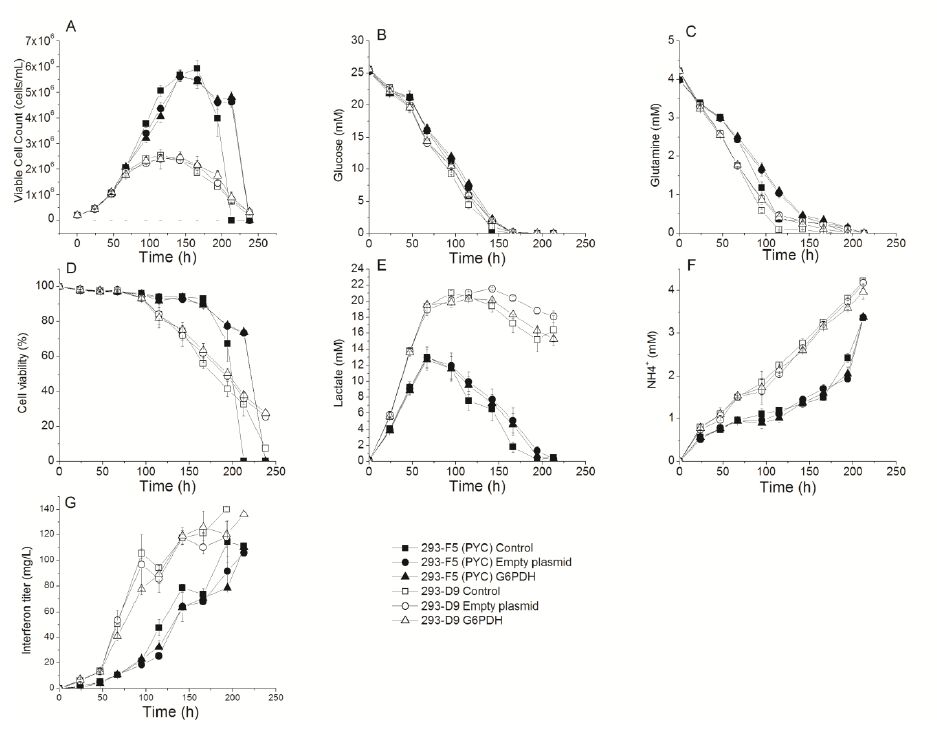
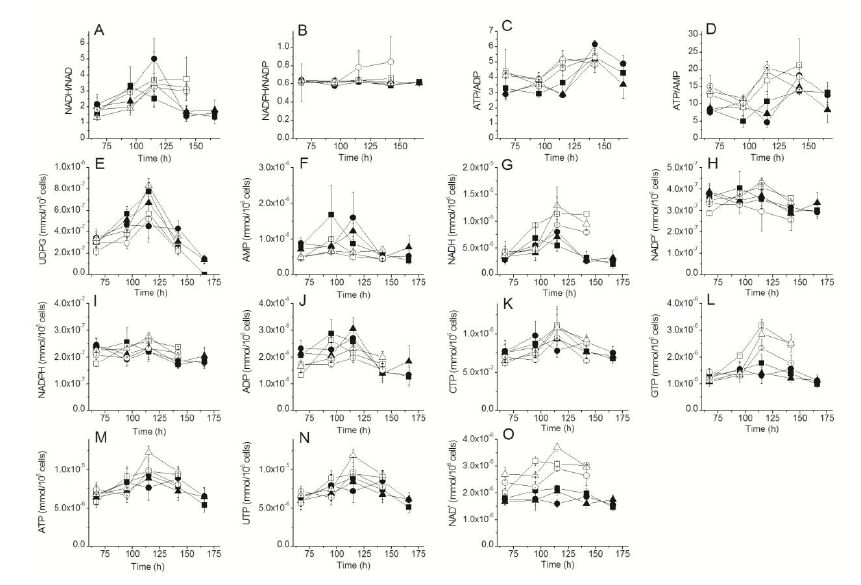
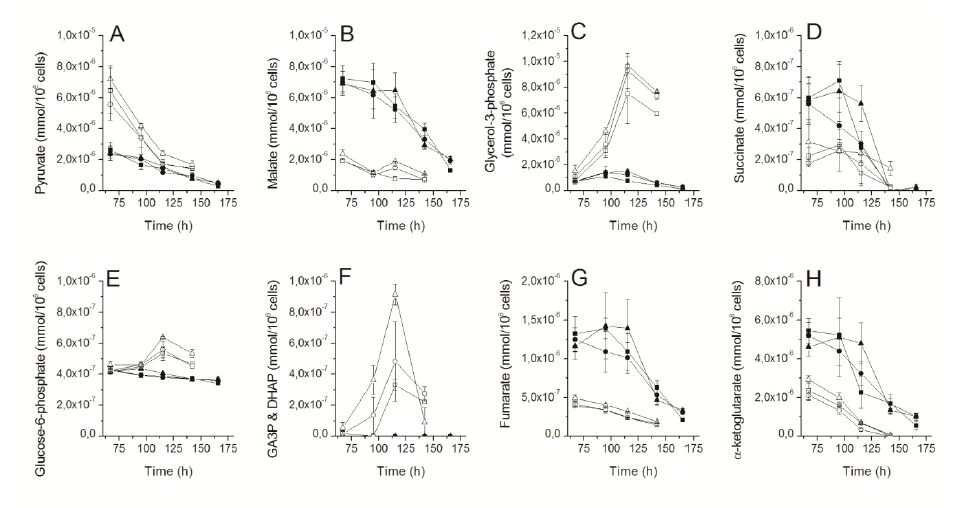
Figures(5)

Edwige Arnold, Iness Hammami, Jingkui Chen, Sachin Gupte, Yves Durocher, Mario Jolicoeur. Overexpression of G6PDH does not affect the behavior of HEK-293 clones stably expressing interferon-α2b[J]. AIMS Bioengineering, 2016, 3(3): 319-336. doi: 10.3934/bioeng.2016.3.319






 DownLoad:
DownLoad: