We aimed to explore key immune-related long non-coding RNAs (lncRNAs) and their effect in predicting of prognosis of triple-negative breast cancer (TNBC).
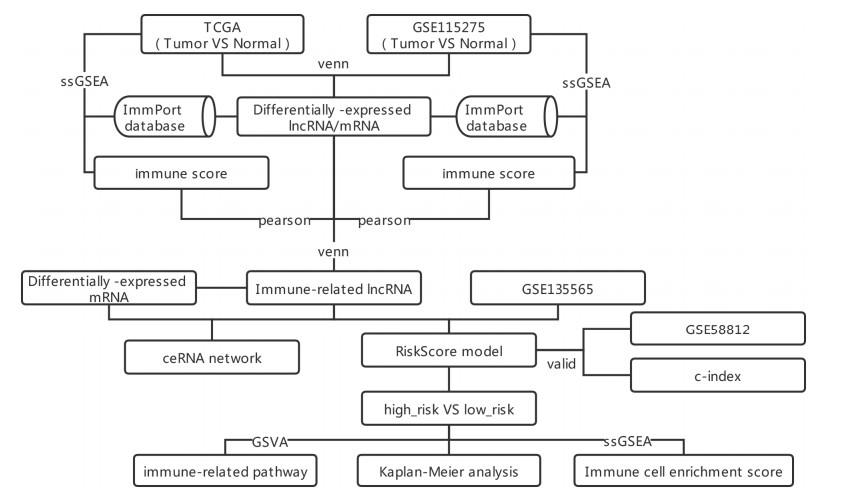
Four datasets of TNBC were downloaded from TCGA and GEO databases. ImmPort database was utilized to acquire immune-related mRNAs. Single sample gene set enrichment analysis (ssGSEA) and correlation analysis were utilized to screen immune-related lncRNAs. Univariate and multivariate Cox regression analyses were utilized to screen independent prognostic lncRNAs to establish prognostic risk model, and the model was evaluated by survival analysis and nomogram. Differential functions and immune cells infiltration in high and low risk group were analyzed by Gene set variation analysis and ssGSEA. Finally, competitive endogenous RNAs was constructed.
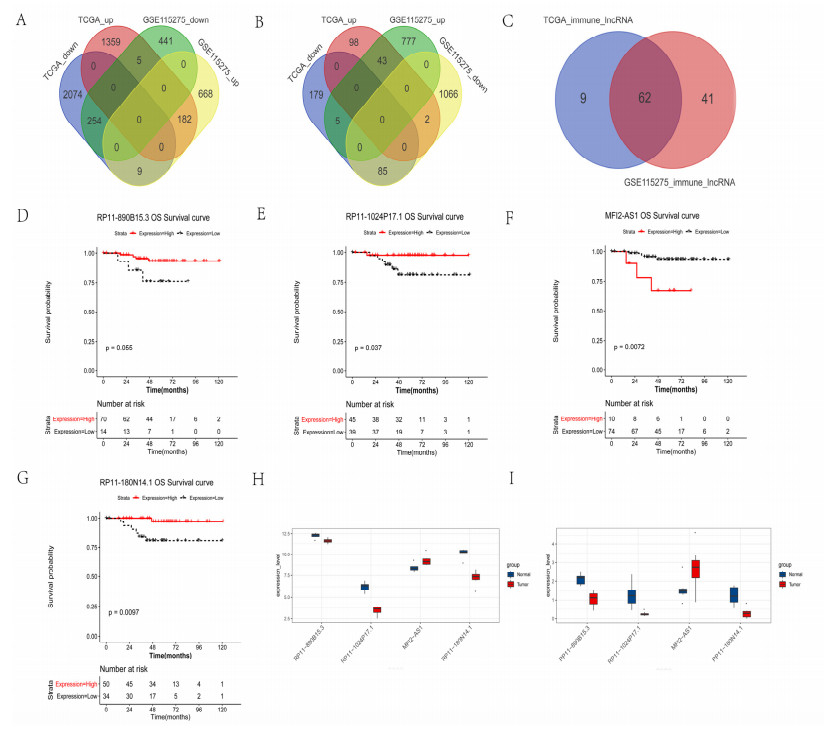
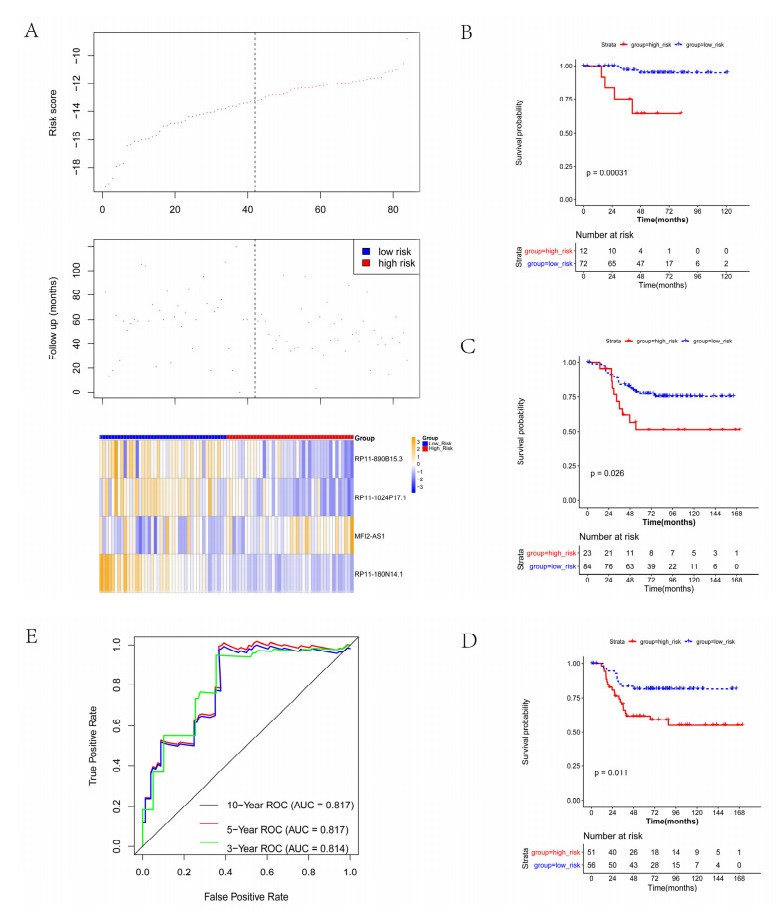
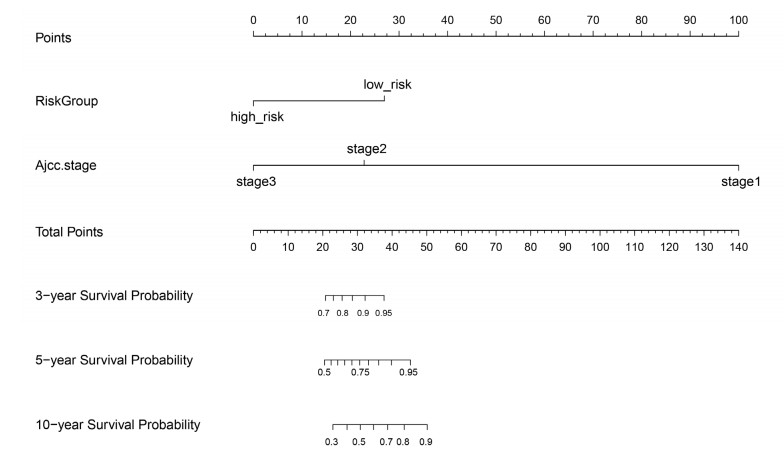
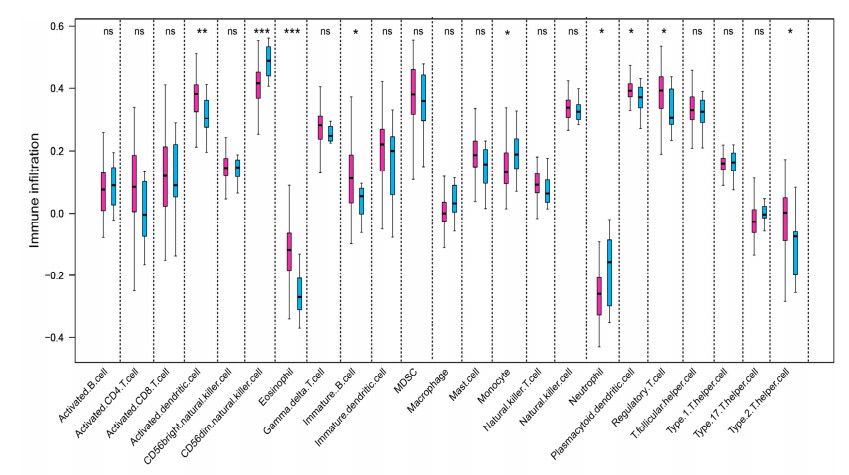
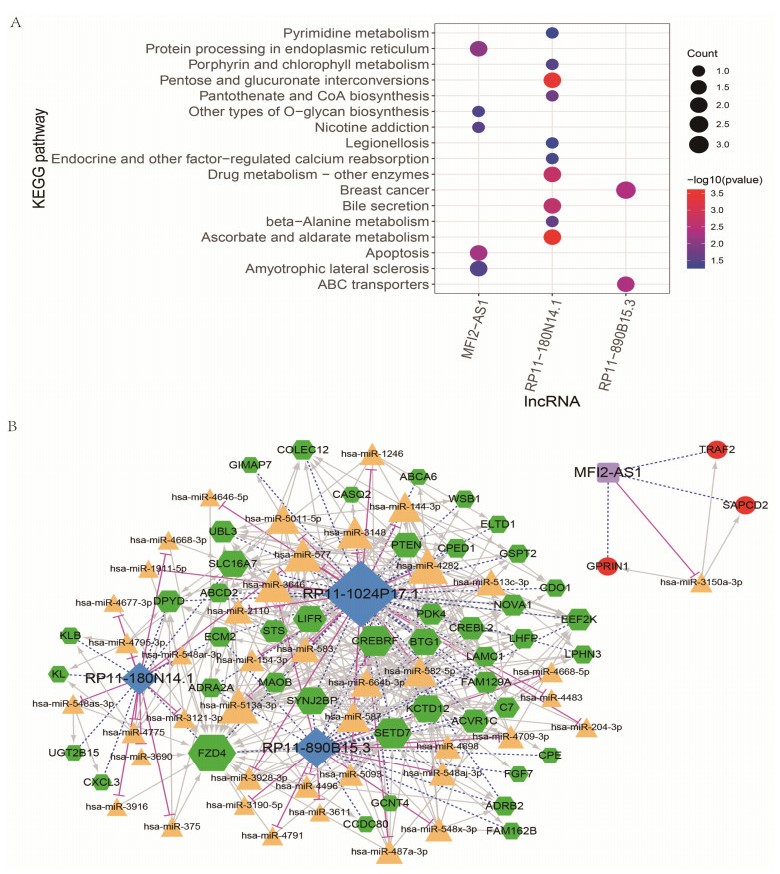
We revealed 62 immune-related lncRNAs, of which four lncRNAs (RP11-890B15.3, RP11-1024P17.1, MFI2-AS1 and RP11-180N14.1) had independent prognostic value. These four lncRNAs-based prognostic risk model could stratify the TNBC patients into high and low risk groups, and patients with high risk displayed unfavorable outcomes. Nomogram indicated that the prognostic model could indicate TNBC patients survival very well. We further found that high risk group showed significantly enriched immune response to tumor cell, humoral immune response and high infiltrating abundance of regulatory T cell, Type 2 T helper cell, eosinophil, etc. LncRNAs RP11-180N14.1, RP11-1024P17.1 and RP11-890B15.3 regulated more mRNAs by targeting various miRNAs. While MFI2-AS1 regulated three mRNAs by sponging miR-3150a-3p.
These four lncRNAs were prognostic biomarkers and could be possible therapeutic targets in TNBC.
Citation: Yun-xiang Li, Shi-ming Wang, Chen-quan Li. Four-lncRNA immune prognostic signature for triple-negative breast cancer[J]. Mathematical Biosciences and Engineering, 2021, 18(4): 3939-3956. doi: 10.3934/mbe.2021197
We aimed to explore key immune-related long non-coding RNAs (lncRNAs) and their effect in predicting of prognosis of triple-negative breast cancer (TNBC).
Four datasets of TNBC were downloaded from TCGA and GEO databases. ImmPort database was utilized to acquire immune-related mRNAs. Single sample gene set enrichment analysis (ssGSEA) and correlation analysis were utilized to screen immune-related lncRNAs. Univariate and multivariate Cox regression analyses were utilized to screen independent prognostic lncRNAs to establish prognostic risk model, and the model was evaluated by survival analysis and nomogram. Differential functions and immune cells infiltration in high and low risk group were analyzed by Gene set variation analysis and ssGSEA. Finally, competitive endogenous RNAs was constructed.
We revealed 62 immune-related lncRNAs, of which four lncRNAs (RP11-890B15.3, RP11-1024P17.1, MFI2-AS1 and RP11-180N14.1) had independent prognostic value. These four lncRNAs-based prognostic risk model could stratify the TNBC patients into high and low risk groups, and patients with high risk displayed unfavorable outcomes. Nomogram indicated that the prognostic model could indicate TNBC patients survival very well. We further found that high risk group showed significantly enriched immune response to tumor cell, humoral immune response and high infiltrating abundance of regulatory T cell, Type 2 T helper cell, eosinophil, etc. LncRNAs RP11-180N14.1, RP11-1024P17.1 and RP11-890B15.3 regulated more mRNAs by targeting various miRNAs. While MFI2-AS1 regulated three mRNAs by sponging miR-3150a-3p.
These four lncRNAs were prognostic biomarkers and could be possible therapeutic targets in TNBC.

| [1] |
A. B. Hanker, D. R. Sudhan, C. L. Arteaga, Overcoming endocrine resistance in breast cancer, Cancer Cell, 37 (2020), 496-513. doi: 10.1016/j.ccell.2020.03.009

|
| [2] |
N. Li, Y. Deng, L. Zhou, T. Tian, S. Yang, Y. Wu, et al., Global burden of breast cancer and attributable risk factors in 195 countries and territories, from 1990 to 2017: results from the Global Burden of Disease Study 2017, J. Hematol. Oncol., 12 (2019), 140. doi: 10.1186/s13045-019-0828-0
|
| [3] |
L. Yin, J. J. Duan, X. W. Bian, S. C. Yu, Triple-negative breast cancer molecular subtyping and treatment progress, Breast Cancer Res., 22 (2020), 61. doi: 10.1186/s13058-020-01296-5
|
| [4] |
M. A. Medina, G. Oza, A. Sharma, L. G. Arriaga, J. M. Hernández, V. M. Rotello, et al., Triple-negative breast cancer: a review of conventional and advanced therapeutic strategies, Int. J. Environ. Res. Public Health, 17 (2020), 2078. doi: 10.3390/ijerph17062078
|
| [5] |
S. Zhao, W. J. Zuo, Z. M. Shao, Y. Z. Jiang, Molecular subtypes and precision treatment of triple-negative breast cancer, Ann. Transl. Med., 8 (2020), 499. doi: 10.21037/atm.2020.03.194
|
| [6] |
T. E. Keenan, S. M. Tolaney, Role of immunotherapy in triple-negative breast cancer, J. Natl. Compr. Cancer Network, 18 (2020), 479-489. doi: 10.6004/jnccn.2020.7554
|
| [7] |
A. Bhan, M. Soleimani, S. S. Mandal, Long noncoding RNA and cancer: a new paradigm, Cancer Res., 77 (2017), 3965-3981. doi: 10.1158/0008-5472.CAN-16-2634
|
| [8] |
W. X. Peng, P. Koirala, Y. Y. Mo, LncRNA-mediated regulation of cell signaling in cancer, Oncogene, 36 (2017), 5661-5667. doi: 10.1038/onc.2017.184
|
| [9] |
W. Wang, W. Lou, B. Ding, B. Yang, H. Lu, Q. Kong, et al., A novel mRNA-miRNA-lncRNA competing endogenous RNA triple sub-network associated with prognosis of pancreatic cancer, Aging, 11 (2019), 2610-2627. doi: 10.18632/aging.101933
|
| [10] | H. Dong, J. Hu, K. Zou, M. Ye, Y. Chen, C. Wu, et al., Activation of LncRNA TINCR by H3K27 acetylation promotes Trastuzumab resistance and epithelial-mesenchymal transition by targeting MicroRNA-125b in breast Cancer, Mol. Cancer, 18 (2019), 3. |
| [11] |
C. N. Fan, L. Ma, N. Liu, Systematic analysis of lncRNA-miRNA-mRNA competing endogenous RNA network identifies four-lncRNA signature as a prognostic biomarker for breast cancer, J. Transl. Med., 16 (2018), 264. doi: 10.1186/s12967-018-1640-2
|
| [12] |
W. Ma, F. Zhao, X. Yu, S. Guan, H. Suo, Z. Tao, et al., Immune-related lncRNAs as predictors of survival in breast cancer: a prognostic signature, J. Transl. Med., 18 (2020), 442. doi: 10.1186/s12967-020-02522-6
|
| [13] |
F. D. E. De Palma, V. Del Monaco, J. G. Pol, M. Kremer, V. D'Argenio, G. Stoll, et al., The abundance of the long intergenic non-coding RNA 01087 differentiates between luminal and triple-negative breast cancers and predicts patient outcome, Pharmacol. Res., 161 (2020), 105249. doi: 10.1016/j.phrs.2020.105249
|
| [14] |
M. Xu, X. Xu, B. Pan, X. Chen, K. Lin, K. Zeng, et al., LncRNA SATB2-AS1 inhibits tumor metastasis and affects the tumor immune cell microenvironment in colorectal cancer by regulating SATB2, Mol. Cancer, 18 (2019), 135. doi: 10.1186/s12943-019-1063-6
|
| [15] |
D. Huang, J. Chen, L. Yang, Q. Ouyang, J. Li, L. Lao, et al., NKILA lncRNA promotes tumor immune evasion by sensitizing T cells to activation-induced cell death, Nat. Immunol., 19 (2018), 1112-1125. doi: 10.1038/s41590-018-0207-y
|
| [16] |
T. Barrett, D. B. Troup, S. E. Wilhite, P. Ledoux, D. Rudnev, C. Evangelista, et al., NCBI GEO: mining tens of millions of expression profiles——database and tools update, Nucleic Acids Res., 35 (2007), D760-765. doi: 10.1093/nar/gkl887
|
| [17] |
J. Harrow, A. Frankish, J. M. Gonzalez, E. Tapanari, M. Diekhans, F. Kokocinski, et al., GENCODE: the reference human genome annotation for The ENCODE Project, Genome Res., 22 (2012), 1760-1774. doi: 10.1101/gr.135350.111
|
| [18] |
H. Jiang, W. H. Wong, SeqMap: mapping massive amount of oligonucleotides to the genome, Bioinformatics, 24 (2008), 2395-2396. doi: 10.1093/bioinformatics/btn429
|
| [19] | G. K. Smyth, limma: Linear Models for Microarray Data, in Bioinformatics and Computational Biology Solutions Using R and Bioconductor, Springer, (2005), 397-420. |
| [20] |
S. Bhattacharya, S. Andorf, L. Gomes, P. Dunn, H. Schaefer, J. Pontius, et al., ImmPort: disseminating data to the public for the future of immunology, Immunol. Res., 58 (2014), 234-239. doi: 10.1007/s12026-014-8516-1
|
| [21] |
D. A. Barbie, P. Tamayo, J. S. Boehm, S. Y. Kim, S. E. Moody, I. F. Dunn, et al., Systematic RNA interference reveals that oncogenic KRAS-driven cancers require TBK1, Nature, 462 (2009), 108-112. doi: 10.1038/nature08460
|
| [22] |
A. Liberzon, A. Subramanian, R. Pinchback, H. Thorvaldsdóttir, P. Tamayo, J. P. Mesirov, Molecular signatures database (MSigDB) 3.0, Bioinformatics, 27 (2011), 1739-1740. doi: 10.1093/bioinformatics/btr260
|
| [23] |
S. Hänzelmann, R. Castelo, J. Guinney, GSVA: gene set variation analysis for microarray and RNA-seq data, BMC Bioinf., 14 (2013), 7. doi: 10.1186/1471-2105-14-7
|
| [24] |
P. Charoentong, F. Finotello, M. Angelova, C. Mayer, M. Efremova, D. Rieder, et al., Pan-cancer immunogenomic analyses reveal genotype-immunophenotype relationships and predictors of response to checkpoint blockade, Cell Rep., 18 (2017), 248-262. doi: 10.1016/j.celrep.2016.12.019
|
| [25] |
G. Yu, L. G. Wang, Y. Han, Q. Y. He, clusterProfiler: an R package for comparing biological themes among gene clusters, Omics, 16 (2012), 284-287. doi: 10.1089/omi.2011.0118
|
| [26] |
M. D. Paraskevopoulou, I. S. Vlachos, D. Karagkouni, G. Georgakilas, I. Kanellos, T. Vergoulis, et al., DIANA-LncBase v2: indexing microRNA targets on non-coding transcripts, Nucleic Acids Res., 44 (2016), D231-238. doi: 10.1093/nar/gkv1270
|
| [27] |
H. Dweep, N. Gretz, miRWalk2.0: a comprehensive atlas of microRNA-target interactions, Nat. Methods, 12 (2015), 697. doi: 10.1038/nmeth.3485
|
| [28] | M. Kohl, S. Wiese, B. Warscheid, Cytoscape: software for visualization and analysis of biological networks, in Data mining in proteomics, Humana Press, (2011), 291-303. |
| [29] |
E. Almagro, C. S. González, E. Espinosa, Prognostic factors of early breast cancer, Med. Clin., 146 (2016), 167-171. doi: 10.1016/j.medcli.2014.12.019
|
| [30] |
P. Costa-Pinheiro, D. Montezuma, R. Henrique, C. Jerónimo, Diagnostic and prognostic epigenetic biomarkers in cancer, Epigenomics, 7 (2015), 1003-1015. doi: 10.2217/epi.15.56
|
| [31] |
M. J. Kwon, Emerging immune gene signatures as prognostic or predictive biomarkers in breast cancer, Arch. Pharm. Res., 42 (2019), 947-961. doi: 10.1007/s12272-019-01189-y
|
| [32] |
Y. Yang, Cancer immunotherapy: harnessing the immune system to battle cancer, J. Clin. Invest., 125 (2015), 3335-3337. doi: 10.1172/JCI83871
|
| [33] |
L. Wang, D. L. Simons, X. Lu, T. Y. Tu, S. Solomon, R. Wang, et al., Connecting blood and intratumoral T(reg) cell activity in predicting future relapse in breast cancer, Nat. Immunol., 20 (2019), 1220-1230. doi: 10.1038/s41590-019-0429-7
|
| [34] |
V. Hashemi, L. A. Maleki, M. Esmaily, A. Masjedi, G. Ghalamfarsa, A. Namdar, et al., Regulatory T cells in breast cancer as a potent anti-cancer therapeutic target, Int. Immunopharmacol., 78 (2020), 106087. doi: 10.1016/j.intimp.2019.106087
|
| [35] |
X. Zhao, J. Liu, S. Ge, C. Chen, S. Li, X. Wu, et al., Saikosaponin a inhibits breast cancer by regulating Th1/Th2 balance, Front. Pharmacol., 10 (2019), 624. doi: 10.3389/fphar.2019.00624
|
| [36] | W. Zhang, Y. Pan, P. Gou, C. Zhou, L. Ma, Q. Liu, et al., Effect of xanthohumol on Th1/Th2 balance in a breast cancer mouse model, Oncol. Rep., 39 (2018), 280-288. |
| [37] |
S. Sakkal, S. Miller, V. Apostolopoulos, K. Nurgali, Eosinophils in cancer: favourable or unfavourable?, Curr. Med. Chem., 23 (2016), 650-666. doi: 10.2174/0929867323666160119094313
|
| [38] | K. Chouliaras, Y. Tokumaru, M. Asaoka, M. Oshi, K. M. Attwood, K. Yoshida, et al., Prevalence and clinical relevance of tumor-associated tissue eosinophilia (TATE) in breast cancer, Surgery, (2020), in press. |
| [39] |
B. Tsuda, A. Miyamoto, K. Yokoyama, R. Ogiya, R. Oshitanai, M. Terao, et al., B-cell populations are expanded in breast cancer patients compared with healthy controls, Breast Cancer, 25 (2018), 284-291. doi: 10.1007/s12282-017-0824-6
|
| [40] |
A. Giro-Perafita, L. Luo, A. Khodadadi-Jamayran, M. Thompson, B. Akgol Oksuz, A. Tsirigos, et al., LncRNA RP11-19E11 is an E2F1 target required for proliferation and survival of basal breast cancer, NPJ Breast Cancer, 6 (2020), 1. doi: 10.1038/s41523-019-0144-4
|
| [41] |
D. Sun, H. Liu, T. Wang, Long noncoding RNA RP11-334E6.12 promotes the proliferation, migration and Invasion of breast cancer cells through the EMT pathway by activating the STAT3 cascade, Cancer Manag. Res, 12 (2020), 1113-1120. doi: 10.2147/CMAR.S237981
|
| [42] |
X. Gao, Y. Lai, Z. Zhang, Y. Ma, Z. Luo, Y. Li, et al., Long Non-coding RNA RP11-480I12.5 promotes the proliferation, migration, and invasion of breast cancer cells through the miR-490-3p-AURKA-Wnt/β-Catenin axis, Front. Oncol., 10 (2020), 948. doi: 10.3389/fonc.2020.00948
|
| [43] | K. Liu, J. Liu, Q. F. Bo, MFI2-AS1 regulates the aggressive phenotypes in glioma by modulating MMP14 via a positive feedback loop, Eur. Rev. Med. Pharmacol. Sci., 23 (2019), 5884-5895. |
| [44] |
T. Yu, L. Tong, Y. Ao, G. Zhang, Y. Liu, H. Zhang, Upregulation of TRIAP1 by the lncRNA MFI2-AS1/miR-125a-5p axis promotes thyroid cancer tumorigenesis, Onco. Targets Ther., 13 (2020), 6967-6974. doi: 10.2147/OTT.S236476
|
| [45] | C. Li, F. Tan, Q. Pei, Z. Zhou, Y. Zhou, L. Zhang, et al., Non-coding RNA MFI2-AS1 promotes colorectal cancer cell proliferation, migration and invasion through miR-574-5p/MYCBP axis, Cell Prolif., 52 (2019), e12632. |
| [46] |
R. Flippot, R. Mouawad, J. P. Spano, M. Rouprêt, E. Compérat, M. O. Bitker, et al., Expression of long non-coding RNA MFI2-AS1 is a strong predictor of recurrence in sporadic localized clear-cell renal cell carcinoma, Sci. Rep., 7 (2017), 8540. doi: 10.1038/s41598-017-08363-6
|
| [47] |
Y. Wei, Z. Wang, Y. Zong, D. Deng, P. Chen, J. Lu, LncRNA MFI2-AS1 promotes HCC progression and metastasis by acting as a competing endogenous RNA of miR-134 to upregulate FOXM1 expression, Biomed. Pharmacother., 125 (2020), 109890. doi: 10.1016/j.biopha.2020.109890
|
| [48] |
R. Luo, J. Song, W. Zhang, L. Ran, Identification of MFI2-AS1, a novel pivotal lncRNA for prognosis of stage Ⅲ/Ⅳ colorectal cancer, Dig. Dis. Sci., 65 (2020), 3538-3550. doi: 10.1007/s10620-020-06064-1
|
| [49] |
C. Jiao, W. Chen, X. Tan, H. Liang, J. Li, H. Yun, et al., Ganoderma lucidum spore oil induces apoptosis of breast cancer cells in vitro and in vivo by activating caspase-3 and caspase-9, J. Ethnopharmacol., 247 (2020), 112256. doi: 10.1016/j.jep.2019.112256
|
| [50] |
Y. Yao, K. Zhao, Z. Yu, H. Ren, L. Zhao, Z. Li, et al., Wogonoside inhibits invasion and migration through suppressing TRAF2/4 expression in breast cancer, J. Exp. Clin. Cancer Res., 36 (2017), 103. doi: 10.1186/s13046-017-0574-5
|
| [51] | Y. Zhang, J. L. Liu, J. Wang, SAPCD2 promotes invasiveness and migration ability of breast cancer cells via YAP/TAZ, Eur. Rev. Med. Pharmacol. Sci., 24 (2020), 3786-3794. |
 mbe-18-04-197 supplementary.zip mbe-18-04-197 supplementary.zip |
 |

Figures(7) / Tables(4)

Yun-xiang Li, Shi-ming Wang, Chen-quan Li. Four-lncRNA immune prognostic signature for triple-negative breast cancer[J]. Mathematical Biosciences and Engineering, 2021, 18(4): 3939-3956. doi: 10.3934/mbe.2021197







 DownLoad:
DownLoad: