Citation: Mohammad Mofatteh. mRNA localization and local translation in neurons[J]. AIMS Neuroscience, 2020, 7(3): 299-310. doi: 10.3934/Neuroscience.2020016

| [1] |
Mofatteh M, Bullock SL (2017) SnapShot: Subcellular mRNA localization. Cell 169: 178-178.e1. doi: 10.1016/j.cell.2017.03.004

|
| [2] |
Holt CE, Schuman EM (2013) The central dogma decentralized: New perspectives on RNA function and local translation in neurons. Neuron 80: 648-657. doi: 10.1016/j.neuron.2013.10.036
|
| [3] |
Buxbaum AR, Haimovich G, Singer RH (2015) In the right place at the right time: visualizing and understanding mRNA localization. Nat Rev Mol Cell Biol 16: 95-109. doi: 10.1038/nrm3918
|
| [4] |
Jeffery WR, Tomlinson CR, Brodeur RD (1983) Localization of actin messenger RNA during early ascidian development. Dev Biol 99: 408-417. doi: 10.1016/0012-1606(83)90290-7
|
| [5] |
Long RM, Singer RH, Meng X, et al. (1997) Mating type switching in yeast controlled by asymmetric localization of ASH1 mRNA. Science 277: 383-387. doi: 10.1126/science.277.5324.383
|
| [6] |
Lawrence JB, Singer RH (1986) Intracellular localization of messenger RNAs for cytoskeletal proteins. Cell 45: 407-415. doi: 10.1016/0092-8674(86)90326-0
|
| [7] | Tabara H, Hill RJ, Mello CC, et al. (1999) pos-1 encodes a cytoplasmic zinc-finger protein essential for germline specification in C. elegans. Development 126: 1-11. |
| [8] |
Berleth T, Burri M, Thoma G, et al. (1988) The role of localization of bicoid RNA in organizing the anterior pattern of the Drosophila embryo. EMBO J 7: 1749-1756. doi: 10.1002/j.1460-2075.1988.tb03004.x
|
| [9] |
Melton DA (1987) Translocation of a localized maternal mRNA to the vegetal pole of Xenopus oocytes. Nature 328: 80-82. doi: 10.1038/328080a0
|
| [10] |
Houle VM, Li W, Montgomery RK, et al. (2003) mRNA localization in polarized intestinal epithelial cells. Am J Physiol Gastrointest Liver Physiol 284: G722-G727. doi: 10.1152/ajpgi.00458.2002
|
| [11] |
Broadus J, Fuerstenberg S, Doe CQ (1998) Staufen-dependent localization of prospero mRNA contributes to neuroblast daughter-cell fate. Nature 391: 792-795. doi: 10.1038/35861
|
| [12] |
Garner CC, Tucker RP, Matus A (1988) Selective localization of messenger RNA for cytoskeletal protein MAP2 in dendrites. Nature 336: 674-677. doi: 10.1038/336674a0
|
| [13] |
Cajigas IJ, Tushev G, Will TJ, et al. (2012) The local transcriptome in the synaptic neuropil revealed by deep sequencing and high-resolution imaging. Neuron 74: 453-466. doi: 10.1016/j.neuron.2012.02.036
|
| [14] |
Köhrmann M, Luo M, Kaether C, et al. (1999) Microtubule-dependent recruitment of Staufen-green fluorescent protein into large RNA-containing granules and subsequent dendritic transport in living hippocampal neurons. Mol Biol Cell 10: 2945-2953. doi: 10.1091/mbc.10.9.2945
|
| [15] |
Mikl M, Vendra G, Kiebler MA (2011) Independent localization of MAP2, CaMKIIα and β-actin RNAs in low copy numbers. EMBO Rep 12: 1077-1084. doi: 10.1038/embor.2011.149
|
| [16] |
Roos J, Hummel T, Ng N, et al. (2000) Drosophila futsch regulates synaptic microtubule organization and is necessary for synaptic growth. Neuron 26: 371-382. doi: 10.1016/S0896-6273(00)81170-8
|
| [17] |
Tiruchinapalli DM, Oleynikov Y, Kelic S, et al. (2003) Activity-dependent trafficking and dynamic localization of zipcode binding protein 1 and beta-actin mRNA in dendrites and spines of hippocampal neurons. J Neurosci 23: 3251-3261. doi: 10.1523/JNEUROSCI.23-08-03251.2003
|
| [18] |
Tongiorgi E, Armellin M, Giulianini PG, et al. (2004) Brain-derived neurotrophic factor mRNA and protein are targeted to discrete dendritic laminas by events that trigger epileptogenesis. J Neurosci 24: 6842-6852. doi: 10.1523/JNEUROSCI.5471-03.2004
|
| [19] |
Park HY, Lim H, Yoon YJ, et al. (2014) Visualization of dynamics of single endogenous mRNA labeled in live mouse. Science 343: 422-424. doi: 10.1126/science.1239200
|
| [20] |
Kanai Y, Dohmae N, Hirokawa N (2004) Kinesin transports RNA: Isolation and characterization of an RNA-Transporting granule. Neuron 43: 513-525. doi: 10.1016/j.neuron.2004.07.022
|
| [21] |
Rook MS, Lu M, Kosik KS (2000) CaMKIIalpha 3′ untranslated region-directed mRNA translocation in living neurons: visualization by GFP linkage. J Neurosci 20: 6385-6393. doi: 10.1523/JNEUROSCI.20-17-06385.2000
|
| [22] |
Ling SC, Fahrner PS, Greenough WT, et al. (2004) Transport of Drosophila fragile X mental retardation protein-containing ribonucleoprotein granules by kinesin-1 and cytoplasmic dynein. Proc Natl Acad Sci 101: 17428-17433. doi: 10.1073/pnas.0408114101
|
| [23] |
Dictenberg JB, Swanger SA, Antar LN, et al. (2008) A direct role for FMRP in activity-dependent dendritic mRNA transport links filopodial-spine morphogenesis to fragile X syndrome. Dev Cell 14: 926-939. doi: 10.1016/j.devcel.2008.04.003
|
| [24] |
Kao DI, Aldridge GM, Weiler IJ, et al. (2010) Altered mRNA transport, docking, and protein translation in neurons lacking fragile X mental retardation protein. Proc Natl Acad Sci 107: 15601-15606. doi: 10.1073/pnas.1010564107
|
| [25] |
Estes PS, O'Shea M, Clasen S, et al. (2008) Fragile X protein controls the efficacy of mRNA transport in Drosophila neurons. Mol Cell Neurosci 39: 170-179. doi: 10.1016/j.mcn.2008.06.012
|
| [26] |
Pan L, Zhang YQ, Woodruff E, et al. (2004) The Drosophila fragile X gene negatively regulates neuronal elaboration and synaptic differentiation. Curr Biol 14: 1863-1870. doi: 10.1016/j.cub.2004.09.085
|
| [27] |
Tucker B, Richards RI, Lardelli M (2006) Contribution of mGluR and Fmr1 functional pathways to neurite morphogenesis, craniofacial development and fragile X syndrome. Hum Mol Genet 15: 3446-3458. doi: 10.1093/hmg/ddl422
|
| [28] |
Dynes JL, Steward O (2007) Dynamics of bidirectional transport of Arc mRNA in neuronal dendrites. J Comp Neurol 500: 433-447. doi: 10.1002/cne.21189
|
| [29] |
Doyle M, Kiebler MA (2011) Mechanisms of dendritic mRNA transport and its role in synaptic tagging. EMBO J 30: 3540-3552. doi: 10.1038/emboj.2011.278
|
| [30] |
Wang C, Han B, Zhou R, et al. (2016) Real-time imaging of translation on single mRNA transcripts in live cells. Cell 165: 990-1001. doi: 10.1016/j.cell.2016.04.040
|
| [31] |
Weatheritt RJ, Gibson TJ, Babu MM (2014) Asymmetric mRNA localization contributes to fidelity and sensitivity of spatially localized systems. Nat Struct Mol Biol 21: 833-839. doi: 10.1038/nsmb.2876
|
| [32] |
Liao G, Mingle L, Van De Water L, et al. (2015) Control of cell migration through mRNA localization and local translation. Wiley Interdiscip Rev RNA 6: 1-15. doi: 10.1002/wrna.1265
|
| [33] |
Park HY, Trcek T, Wells AL, et al. (2012) An unbiased analysis method to quantify mRNA localization reveals its correlation with cell motility. Cell Rep 1: 179-184. doi: 10.1016/j.celrep.2011.12.009
|
| [34] |
Twiss JL, Fainzilber M (2009) Ribosomes in axons- scrounging from the neighbors? Trends Cell Biol 19: 236-243. doi: 10.1016/j.tcb.2009.02.007
|
| [35] |
Eng H, Lund K, Campenot RB (1999) Synthesis of β-Tubulin, actin, and other proteins in axons of sympathetic neurons in compartmented cultures. J Neurosci 19: 1-9. doi: 10.1523/JNEUROSCI.19-01-00001.1999
|
| [36] |
Giuditta A, Menichini E, Capano CP, et al. (1991) Active polysomes in the axoplasm of the squid giant axon. J Neurosci Res 28: 18-28. doi: 10.1002/jnr.490280103
|
| [37] |
Bassell GJ, Zhang H, Byrd AL, et al. (1998) Sorting of beta-actin mRNA and protein to neurites and growth cones in culture. J Neurosci 18: 251-265. doi: 10.1523/JNEUROSCI.18-01-00251.1998
|
| [38] |
Bunge MB (1973) Fine structure of nerve fibers and growth cones of isolated sympathetic neurons in culture. J Cell Biol 56: 713-735. doi: 10.1083/jcb.56.3.713
|
| [39] |
Jung H, Yoon BC, Holt CE (2012) Axonal mRNA localization and local protein synthesis in nervous system assembly, maintenance and repair. Nat Rev Neurosci 13: 308-324. doi: 10.1038/nrn3210
|
| [40] |
Shigeoka T, Jung H, Jung J, et al. (2016) Dynamic axonal translation in developing and mature visual circuits. Cell 166: 181-192. doi: 10.1016/j.cell.2016.05.029
|
| [41] |
Tcherkezian J, Brittis PA, Thomas F, et al. (2010) Transmembrane receptor DCC associates with protein synthesis machinery and regulates translation. Cell 141: 632-644. doi: 10.1016/j.cell.2010.04.008
|
| [42] |
Koppers M, Cagnetta R, Shigeoka T, et al. (2019) Receptor-specific interactome as a hub for rapid cue-induced selective translation in axons. eLife 8: e48718. doi: 10.7554/eLife.48718
|
| [43] |
Zheng JQ, Kelly TK, Chang B, et al. (2001) A functional role for intra-axonal protein synthesis during axonal regeneration from adult sensory neurons. J Neurosci 21: 9291-9303. doi: 10.1523/JNEUROSCI.21-23-09291.2001
|
| [44] |
Merianda TT, Lin AC, Lam JSY, et al. (2009) A functional equivalent of endoplasmic reticulum and Golgi in axons for secretion of locally synthesized proteins. Mol Cell Neurosci 40: 128-142. doi: 10.1016/j.mcn.2008.09.008
|
| [45] |
Spencer GE, Syed NI, van Kesteren E, et al. (2000) Synthesis and functional integration of a neurotransmitter receptor in isolated invertebrate axons. J Neurobiol 44: 72-81. doi: 10.1002/1097-4695(200007)44:1<72::AID-NEU7>3.0.CO;2-#
|
| [46] |
Cagnetta R, Frese CK, Shigeoka T, et al. (2018) Rapid cue-specific remodeling of the nascent axonal proteome. Neuron 99: 29-46. e4. doi: 10.1016/j.neuron.2018.06.004
|
| [47] |
Verma P, Chierzi S, Codd AM, et al. (2005) Axonal protein synthesis and degradation are necessary for efficient growth cone regeneration. J Neurosci 25: 331-342. doi: 10.1523/JNEUROSCI.3073-04.2005
|
| [48] |
Campbell DS, Holt CE (2001) Chemotropic responses of retinal growth cones mediated by rapid local protein synthesis and degradation. Neuron 32: 1013-1026. doi: 10.1016/S0896-6273(01)00551-7
|
| [49] |
Andreassi C, Zimmermann C, Mitter R, et al. (2010) An NGF-responsive element targets myo-inositol monophosphatase-1 mRNA to sympathetic neuron axons. Nat Neurosci 13: 291-301. doi: 10.1038/nn.2486
|
| [50] |
Zivraj KH, Tung YCL, Piper M, et al. (2010) Subcellular profiling reveals distinct and developmentally regulated repertoire of growth cone mRNAs. J Neurosci 30: 15464-15478. doi: 10.1523/JNEUROSCI.1800-10.2010
|
| [51] |
Lin AC, Holt CE (2007) Local translation and directional steering in axons. EMBO J 26: 3729-3736. doi: 10.1038/sj.emboj.7601808
|
| [52] | Harris WA, Holt CE, Bonhoeffer F (1987) Retinal axons with and without their somata, growing to and arborizing in the tectum of Xenopus embryos: a time-lapse video study of single fibres in vivo. Development 101: 123-133. |
| [53] |
Sahoo PK, Smith DS, Perrone-Bizzozero N, et al. (2018) Axonal mRNA transport and translation at a glance. J Cell Sci 131: jcs196808. doi: 10.1242/jcs.196808
|
| [54] |
Cosker KE, Fenstermacher SJ, Pazyra-Murphy MF, et al. (2016) The RNA-binding protein SFPQ orchestrates an RNA regulon to promote axon viability. Nat Neurosci 19: 690-696. doi: 10.1038/nn.4280
|
| [55] |
Sasaki Y, Welshhans K, Wen Z, et al. (2010) Phosphorylation of zipcode binding protein 1 is required for brain-derived neurotrophic factor signaling of local beta-actin synthesis and growth cone turning. J Neurosci 30: 9349-9358. doi: 10.1523/JNEUROSCI.0499-10.2010
|
| [56] |
Spillane M, Ketschek A, Donnelly CJ, et al. (2012) Nerve growth factor-induced formation of axonal filopodia and collateral branches involves the intra-axonal synthesis of regulators of the actin-nucleating Arp2/3 complex. J Neurosci 32: 17671-17689. doi: 10.1523/JNEUROSCI.1079-12.2012
|
| [57] |
Wong HHW, Lin JQ, Ströhl F, et al. (2017) RNA docking and local translation regulate site-specific axon remodeling in vivo. Neuron 95: 852-868. e8. doi: 10.1016/j.neuron.2017.07.016
|
| [58] |
Mardakheh FK, Paul A, Kümper S, et al. (2015) Global analysis of mRNA, translation, and protein localization: Local translation is a key regulator of cell protrusions. Dev Cell 35: 344-357. doi: 10.1016/j.devcel.2015.10.005
|
| [59] |
Shigeoka T, Jung H, Jung J, et al. (2016) Dynamic axonal translation in developing and mature visual circuits. Cell 166: 181-192. doi: 10.1016/j.cell.2016.05.029
|
| [60] |
Piper M, Anderson R, Dwivedy A, et al. (2006) Signaling mechanisms underlying Slit2-induced collapse of Xenopus retinal growth cones. Neuron 49: 215-228. doi: 10.1016/j.neuron.2005.12.008
|
| [61] |
Meyer MP, Smith SJ (2006) Evidence from in vivo imaging that synaptogenesis guides the growth and branching of axonal arbors by two distinct mechanisms. J Neurosci 26: 3604-3614. doi: 10.1523/JNEUROSCI.0223-06.2006
|
| [62] |
Sanchez G, Dury AY, Murray LM, et al. (2012) A novel function for the survival motoneuron protein as a translational regulator. Hum Mol Genet 22: 668-684. doi: 10.1093/hmg/dds474
|
| [63] |
Shukla S, Parker R (2016) Hypo- and Hyper-Assembly diseases of RNA-Protein complexes. Trends Mol Med 22: 615-628. doi: 10.1016/j.molmed.2016.05.005
|
| [64] |
Jung H, Yoon BC, Holt CE (2012) Axonal mRNA localization and local protein synthesis in nervous system assembly, maintenance and repair. Nat Rev Neurosci 13: 308-324. doi: 10.1038/nrn3210
|
| [65] |
Hao le T, Duy PQ, An M, et al. (2017) HuD and the survival motor neuron protein interact in motoneurons and are essential for motoneuron development, function, and mRNA regulation. J Neurosci 37: 11559-11571. doi: 10.1523/JNEUROSCI.1528-17.2017
|
| [66] |
Fallini C, Zhang H, Su Y, et al. (2011) The survival of motor neuron (SMN) protein interacts with the mRNA-binding protein HuD and regulates localization of poly(A) mRNA in primary motor neuron axons. J Neurosci 31: 3914-3925. doi: 10.1523/JNEUROSCI.3631-10.2011
|
| [67] |
Akten B, Kye MJ, Hao LT, et al. (2011) Interaction of survival of motor neuron (SMN) and HuD proteins with mRNA cpg15 rescues motor neuron axonal deficits. Proc Natl Acad Sci 108: 10337-10342. doi: 10.1073/pnas.1104928108
|
| [68] |
Fallini C, Donlin-Asp PG, Rouanet JP, et al. (2016) Deficiency of the survival of motor neuron protein impairs mRNA localization and local translation in the growth cone of motor neurons. J Neurosci 36: 3811-3820. doi: 10.1523/JNEUROSCI.2396-15.2016
|
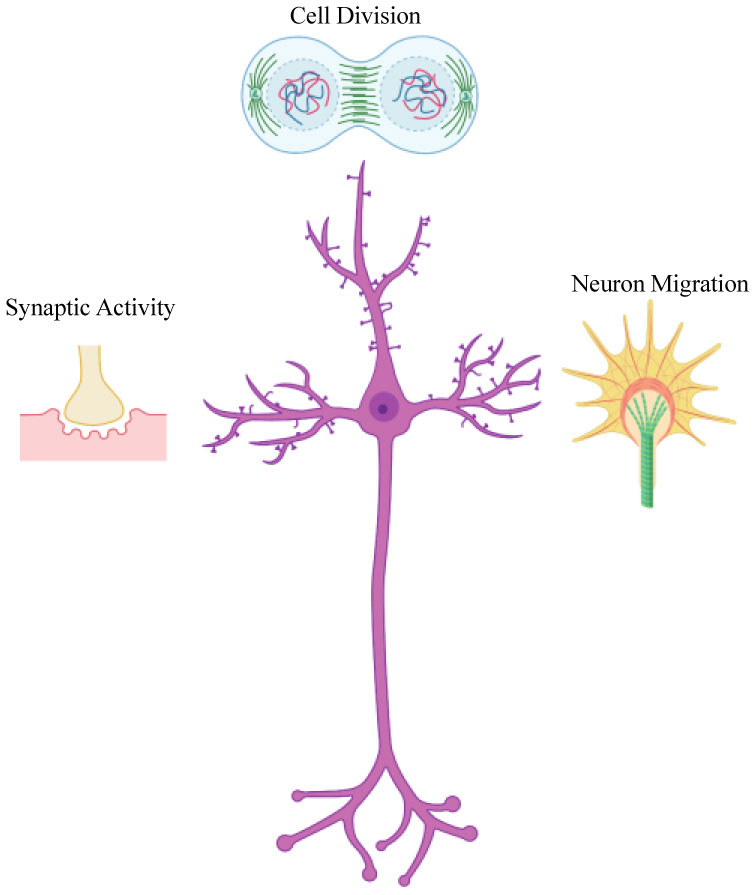
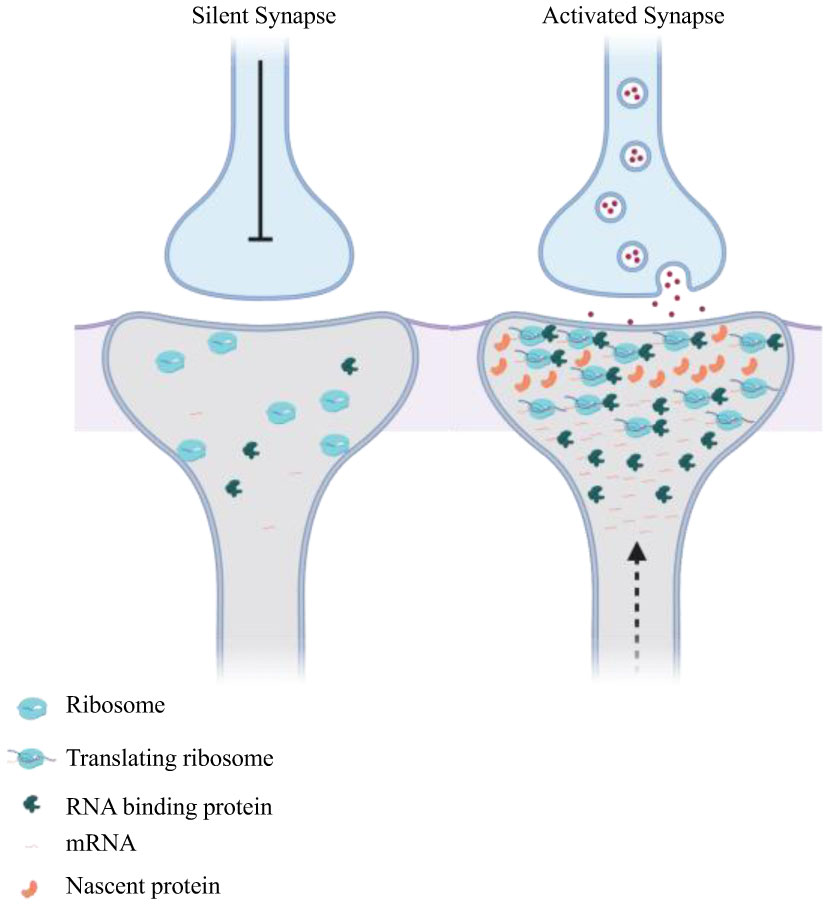
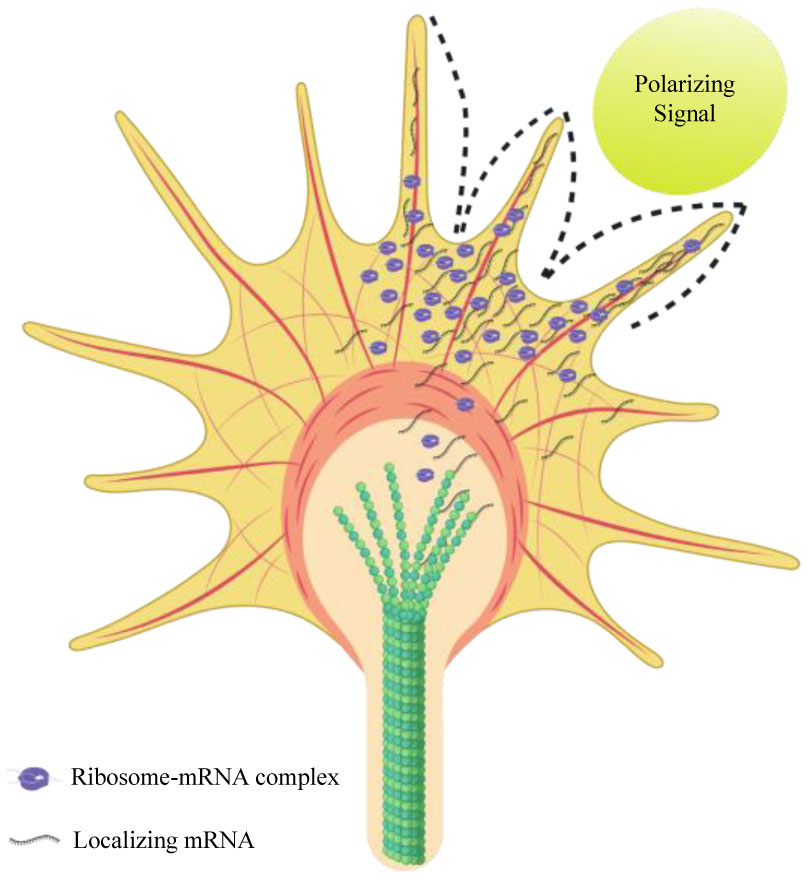
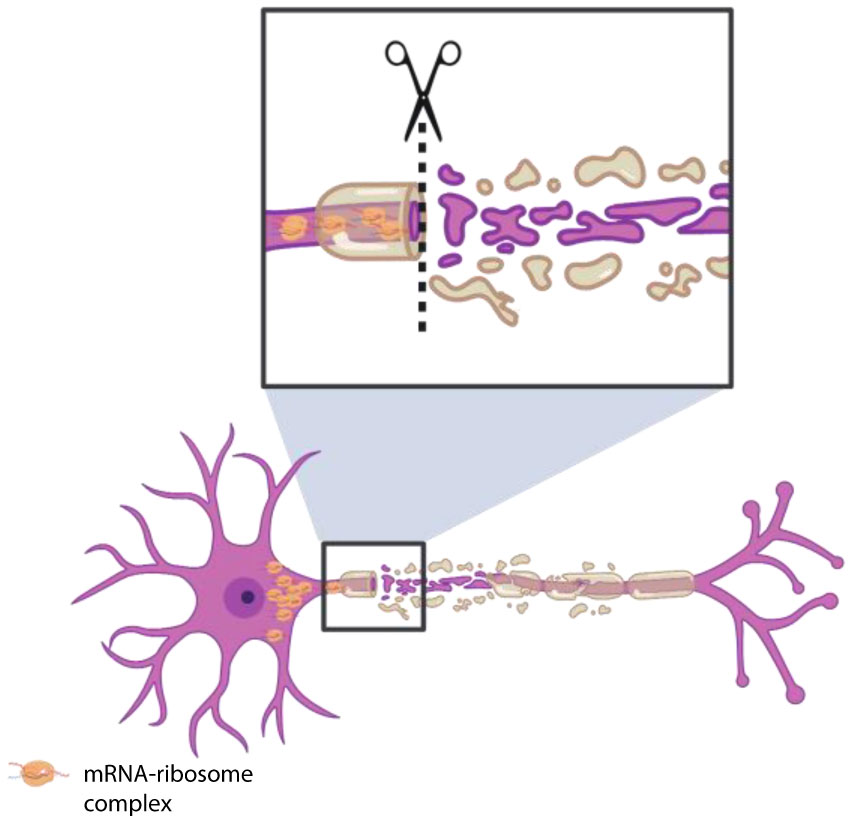
Figures(4)

Mohammad Mofatteh. mRNA localization and local translation in neurons[J]. AIMS Neuroscience, 2020, 7(3): 299-310. doi: 10.3934/Neuroscience.2020016







 DownLoad:
DownLoad: