Citation: M. Laura Donnet, Iraís Dámaris López Becerril, J. Roy Black, Jon Hellin. Productivity differences and food security: a metafrontier analysis of rain-fed maize farmers in MasAgro in Mexico[J]. AIMS Agriculture and Food, 2017, 2(2): 129-148. doi: 10.3934/agrfood.2017.2.129

| [1] |
Eakin H, Bausch JC, Sweeney S (2014) Agrarian Winners of Neoliberal Reform: The 'Maize Boom' of Sinaloa, Mexico. J Agrar Chang 14: 26-51. doi: 10.1111/joac.12005

|
| [2] |
Sweeney S, Steigerwald D, Davenport F, et al. (2013) Mexican Maize Production: Evolving Organizational and Spatial Structures since 1980. Appl Geogr 39: 78-92. doi: 10.1016/j.apgeog.2012.12.005
|
| [3] |
Camacho-Villa TC, Almekinders C, Hellin J, et al. (2016) The evolution of the MasAgro hubs: responsiveness and serendipity as drivers of agricultural innovation in a dynamic and heterogeneous context. J Agric Educ Ext 22: 455-470. doi: 10.1080/1389224X.2016.1227091
|
| [4] |
Appendini K (2014) Reconstructing the Maize Market in Rural Mexico. J Agrar Chang 14: 1-25. doi: 10.1111/joac.12013
|
| [5] |
Ali M, Byerlee D (1991) Economic efficiency of small farmers in a changing world: a survey of recent evidence. J Int Dev 3: 1-27. doi: 10.1002/jid.4010030102
|
| [6] | Fan S, Brzeska J, Keyzer M, et al. (2013) From Subsistence to Profit Transforming Smallholder Farms. International Food Policy Research Institute Washington, DC. |
| [7] |
O'Donnell CJ, Rao DSP, Battese GE (2008) Metafrontier frameworks for the study of firm-level efficiencies and technology ratios. Empir Econ 34: 231-255. doi: 10.1007/s00181-007-0119-4
|
| [8] | Yunez-Naude A, Juarez-Torres M, Barceinas-Paredes F (2006) Productive Efficiency in Agriculture: Corn Production in Mexico. 2006 Annual Meeting, August 12-18, 2006, Queensland, Australia. |
| [9] | Kagin J, Taylor E, Yúnez-Naude A (2015) Inverse productivity or inverse efficiency?: Evidence from Mexico. J Dev Stud 52: 396-411. |
| [10] |
Villano R, Bravo-Ureta B, Solís D, et al. (2015) Modern Rice Technologies and Productivity in the Philippines: Disentangling Technology from Managerial Gaps. J Agric Econ 66: 129-154. doi: 10.1111/1477-9552.12081
|
| [11] | Villano R, Fleming P, Fleming E (2008) Measuring Regional Productivity Differences in the Australian Wool Industry: A Metafrontier Approach. AARES 52nd Annual Conference. |
| [12] |
Kramol P, Villano R, Kristiansen P, et al. (2015) Productivity differences between organic and other vegetable farming systems in northern Thailand. Renew Agric Food Syst 30: 154-169. doi: 10.1017/S1742170513000288
|
| [13] | Turrent A, Wise T, Garvey E (2012) Achieving Mexico's Maize Potential. GDAE Working Paper 12-03. |
| [14] | DOF, Diario Oficial de la Federacion. Programa Sectorial de Desarrollo Agropecuario, Pesquero y Alimentario 20132018. 2013, Available from: https://www.gob.mx/cms/uploads/attachment/file/82434/DOF_-_Diario_Oficial_de_la_Federaci_n.pdf |
| [15] | FAO, IFAD, WFP (2015) The State of Food Insecurity in the World 2015. Meeting the 2015 international hunger targets: taking stock of uneven progress. Rome, FAO. |
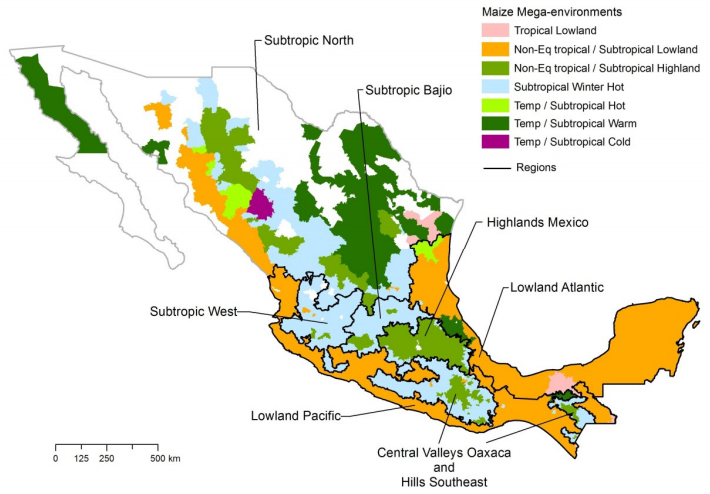
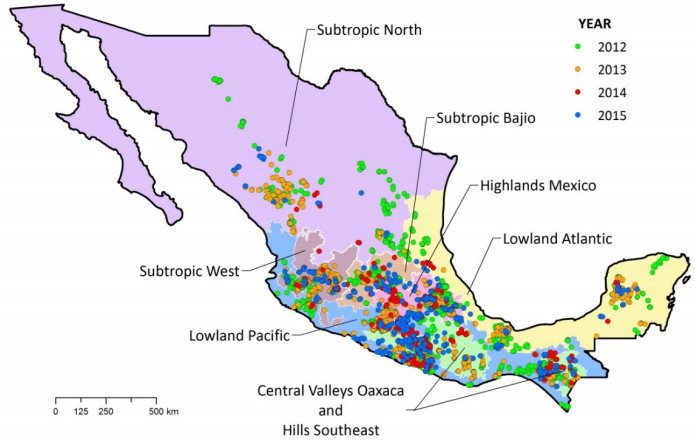
| [16] | Hartkamp AD, White JW, Rodríguez Aguilar A, et al. (2000) Maize Production Environments Revisited: A GIS-based Approach. CIMMYT, Mexico D.F. |
| [17] | Fischer R, Byerlee D, Edmeades G (2014) Crop yields and global food security: will yield increase continue to feed the world? Canberra: Australian Centre for International Agricultural Research. |
| [18] | SIAP, Sistema de Información Agrícola y Pecuaria. Anuario Estadístico de la Producción Agrícola 2012. 2013, Available from: http://infosiap.siap.gob.mx/aagricola_siap/icultivo/index.jsp |
| [19] | SIAP, Sistema de Información Agrícola y Pecuaria. Anuario Estadístico de la Producción Agrícola 2012 - 2015. 2016, Available from: http://infosiap.siap.gob.mx/aagricola_siap/icultivo/index.jsp |
| [20] | INEGI, Instituto Nacional de Estadística y Geografía. Continuo de Elevaciones Mexicano 2.0. Marco Geoestadístico Nacional MGM. 2014, Available from: http://www.inegi.org.mx/geo/contenidos/datosrelieve/continental/continuoelevaciones.aspx |
| [21] | SMN, Servicio Meteorológico Nacional (2015) Resúmenes Mensuales de Temperaturas y Lluvia. |
| [22] |
Aigner D, Lovell C, Schmidt P (1977) Formulation and estimation of stochastic frontier production function models. J Econom 6:21-37. doi: 10.1016/0304-4076(77)90052-5
|
| [23] |
Battese GE (1992) Frontier production functions and technical efficiency: a survey of empirical applications in agricultural economics. Agric Econ 7: 185-208. doi: 10.1016/0169-5150(92)90049-5
|
| [24] | Greene WH (2008) The Econometric Approach to Efficiency Analysis. In: The Measurement of Productive Efficiency and Productivity Change. Oxford University Press. |
| [25] | Namonje-Kapembwa T, Black R, Jayne TS (2015) Does Late Delivery of Subsidized Fertilizer Affect Smallholder Maize Productivity and Production? 2015 AAEA & WAEA Joint Annual Meeting, July 26-28, San Francisco, California 205288, Agricultural and Applied Economics Association; Western Agricultural Economics Association. |
| [26] | Dadzie S, Dasmani I (2010) Gender difference and farm level efficiency: Meta-frontier production function approach. J Dev Agric Econ 2: 441-451. |
| [27] | Hayami Y, Ruttan V (1970) Agricultural productivity differences among countries. Am Econ Rev 60: 895-911. |
| [28] | Mehrabi Boshrabadi H, Villano R, Fleming E (2008) Technical efficiency and environmental - technological gaps in wheat production in Kerman Province of Iran: A meta-frontier analysis. Agric Econ 38: 67-76. |
| [29] |
Mariano M, Villano R, Fleming E (2011) Technical efficiency of rice farms in different agroclimatic zones in the Philippines: An application of a stochastic metafrontier model. Asian Econ J 25: 245-269. doi: 10.1111/j.1467-8381.2011.02060.x
|
| [30] |
Bravo-Ureta B, Greene W, Solis D (2012) Technical Efficiency Analysis Correcting for the Biases from Observed and Unobserved Variables: An Application to a Natural Resource Management Project. Empir Econ 43: 55-72. doi: 10.1007/s00181-011-0491-y
|
| [31] | Kunbhaker S, Wang H, Horncastle A (2015) A Practitioner's Guide to Stochastic Frontier Analysis Using Stata, Cambridge University Press. |
Figures(2) / Tables(7)

M. Laura Donnet, Iraís Dámaris López Becerril, J. Roy Black, Jon Hellin. Productivity differences and food security: a metafrontier analysis of rain-fed maize farmers in MasAgro in Mexico[J]. AIMS Agriculture and Food, 2017, 2(2): 129-148. doi: 10.3934/agrfood.2017.2.129







 DownLoad:
DownLoad: