




Citation: Fatima Al-Hashimi, Salvador J. Diaz-Cano. Multi-target analysis of neoplasms for the evaluation of tumor progression: stochastic approach of biologic processes[J]. AIMS Molecular Science, 2018, 5(1): 14-62. doi: 10.3934/molsci.2018.1.14
| [1] | Khushboo Desai, Dolly Patel, Parth Desai, Rakesh Rawal, Himanshu Pandya . Periostin – an unexplored tumor marker of oral squamous cell carcinoma. AIMS Molecular Science, 2020, 7(4): 383-395. doi: 10.3934/molsci.2020019 |
| [2] | Chunzheng Li, Chenyu Wei, Gongke Zhao, Xianguang Yang . Cancer cells remodeling and quality control are inextricably linked to autophagy. AIMS Molecular Science, 2023, 10(2): 109-126. doi: 10.3934/molsci.2023009 |
| [3] | Simone Leggeri, Navid Sobhani . Single nucleotide polymorphisms Rs1045642 C>T genetic alteration in ATP Binding Cassette Subfamily B Member 1 role in increasing everolimus toxicity in metastatic breast cancer. AIMS Molecular Science, 2020, 7(1): 1-11. doi: 10.3934/molsci.2020001 |
| [4] | Karen E. Livermore, Jennifer Munkley, David J. Elliott . Androgen receptor and prostate cancer. AIMS Molecular Science, 2016, 3(2): 280-299. doi: 10.3934/molsci.2016.2.280 |
| [5] | Uday Chintapula, Samir M Iqbal, Young-Tae Kim . A compendium of single cell analysis in aging and disease. AIMS Molecular Science, 2020, 7(1): 49-69. doi: 10.3934/molsci.2020004 |
| [6] | Sreemita Majumdar, Song-Tao Liu . Cell division symmetry control and cancer stem cells. AIMS Molecular Science, 2020, 7(2): 82-101. doi: 10.3934/molsci.2020006 |
| [7] | Dora Brites . Cell ageing: a flourishing field for neurodegenerative diseases. AIMS Molecular Science, 2015, 2(3): 225-258. doi: 10.3934/molsci.2015.3.225 |
| [8] | Hannah P. Priyanka, Rahul S. Nair, Sanjana Kumaraguru, Kirtikesav Saravanaraj, Vasantharekha Ramasamy . Insights on neuroendocrine regulation of immune mediators in female reproductive aging and cancer. AIMS Molecular Science, 2021, 8(2): 127-148. doi: 10.3934/molsci.2021010 |
| [9] | Xin Nian, Yasuhiro Nagai, Cameron Jeffers, Kara N. Maxwell, Hongtao Zhang . Dietary influence on estrogens and cytokines in breast cancer. AIMS Molecular Science, 2017, 4(3): 252-270. doi: 10.3934/molsci.2017.3.252 |
| [10] | Vahid Pouresmaeil, Marwa Mawlood Salman Al-zand, Aida Pouresmaeil, Seyedeh Samira Saghravanian, Masoud Homayouni Tabrizi . Loading diltiazem onto surface-modified nanostructured lipid carriers to evaluate its apoptotic, cytotoxic, and inflammatory effects on human breast cancer cells. AIMS Molecular Science, 2024, 11(3): 231-250. doi: 10.3934/molsci.2024014 |
Our current general definition of neoplasm ("cellular disease characterized by abnormal growth regulatory mechanisms") is descriptive and challenging to apply routinely, therefore requiring working definitions. Biologically, neoplasms develop through the acquisition of capabilities that involve tumor cell aspects and modified microenvironment interactions, resulting in unlimited growth due to a stepwise accumulation of cooperative genetic alterations that affect critical molecular pathways. The correlation of these molecular aspects with morphological changes is essential for better understanding of fundamental concepts as early neoplasms/precancerous lesions, progression/blocked differentiation, and intratumor heterogeneity [1,2]. The acquired capabilities include self-maintained replication (cell cycle dysregulation), extended cell survival (cell cycle arrest, apoptosis dysregulation, and replicative lifespan), genetic instability (chromosomal and microsatellite), changes in chromatin, transcription and epigenetics, mobilization of cellular resources, and modified microenvironment interactions (tumour cells, stromal cells, extracellular, endothelium). These acquired capabilities defining neoplasms are the hallmarks of cancer, but they also comprise useful tools to improve diagnosis and prognosis, as well as potential therapeutic targets. The introduction of new markers has improved the diagnostic precision, but can potentially result in significant changes in prevalence and uncertainties for particular lesions. The current WHO classifications of tumors incorporate new developments based on pathology and genetics, the leading criteria still being morphological: Molecular findings complement the histological evaluation without replacing it. The acquired capabilities of neoplasms include tumor cell aspects (self-maintained replication, more prolonged cell survival, genetic instability), and the tumor cell microenvironment interaction (induction of neoangiogenesis, invasion, and metastasis) [3].
Considering tumor progression from a biologic perspective, it represents the acquisition of new capabilities relevant for a neoplasm to extend: Invasion of the underlying stroma for intraepithelial lesions and the metastatic potential for already invasive neoplasms. The core mechanisms involved in this process would require maintaining the cellular survival, and proliferation capacity for expansion, along with invasion and neoangiogenesis. A proper evaluation would then require the analysis of at least two samples taken at different times to assess the differences in the acquired capabilities of invasion and metastatic potential; however, the question of tumor progression arises in cases for which only one sample is available. In this context, the most reliable results would be provided by multi-target gene analysis at different structural levels (DNA/RNA/proteins) targeting core cellular functions promoting neoplasms (cellular survival, proliferation capacity, invasion, and cytostasis-differentiation) that result from the interaction of tumor cells and their microenvironment (Figure 1).
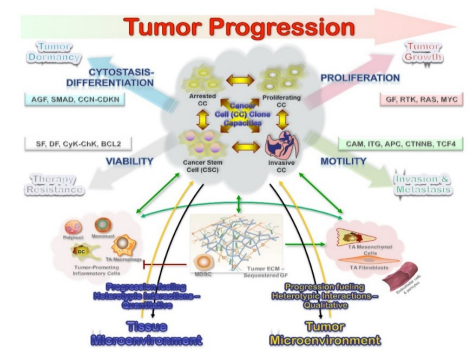 Figure 1. As we do not know when the tumor started and the sequence of genetic alterations, their interactions, and interactive effects, a stochastic approach would be the most useful. In this context, the concept of a constellation of genes or gene clusters at the core of tumor development is the most appropriate. Tumor progression is not a linear process; it requires maintaining the proliferation capacity for expansion and invasion, along with cellular survival and cytostasis-differentiation. These four principal mechanisms result from the interaction between the tumor cells and the balance between the tumor-promoting and inhibiting signals in the tissue. The tissue microenvironment is thought to play a significant role, as the tumor-associated (TA) stromal cells and macrophages induce or up-regulate the production of several pro-malignancy factors from the tumor cells.
Figure 1. As we do not know when the tumor started and the sequence of genetic alterations, their interactions, and interactive effects, a stochastic approach would be the most useful. In this context, the concept of a constellation of genes or gene clusters at the core of tumor development is the most appropriate. Tumor progression is not a linear process; it requires maintaining the proliferation capacity for expansion and invasion, along with cellular survival and cytostasis-differentiation. These four principal mechanisms result from the interaction between the tumor cells and the balance between the tumor-promoting and inhibiting signals in the tissue. The tissue microenvironment is thought to play a significant role, as the tumor-associated (TA) stromal cells and macrophages induce or up-regulate the production of several pro-malignancy factors from the tumor cells.The development of clinically detectable tumors requires the accumulation of some cooperative genetic alterations, regardless of its order [4]. The average number of core genetic alterations requires for neoplastic transformation should reflect the number of essential cellular processes involved in tumorigenesis (we have highlighted the four core mechanisms in this review). As the biological mechanisms are redundant, there will be mutations in more than one gene. The genomic tumor analyses demonstrate a myriad of these genetic alterations, but not all of them are driver mutations. At the very least, to bypass the biologic redundancy, it should be assumed at least two genetic changes in more than 50% of these biological processes. As such, tan averge of 6–7 genetic alterations are required for clinically detectable tumors, correlating with both morphological progression in some locations and intratumor heterogeneity. These capabilities are not equally relevant at different stages during tumorigenesis, as highlighted by careful morphological evaluations. Intratumor heterogeneity in human tumors is a widespread phenomenon of critical importance for tumor progression and the response to therapeutic intervention, and it is a crucial variable to understand tumor natural history and potential response to therapy. It is inherent to neoplasms from early stages and is also the byproduct of tumor progression as genetic abnormalities accumulate. It usually has been assumed that tumor progression is a linear process, being metastasis a late event. However, this model would not match well with the heterogeneity: The invasive capability can be early acquired and result in metastasis from early neoplasms that already show genetic and kinetic features of established malignancies, even for those of low nuclear grade. Human cancers frequently display substantial intratumor heterogeneity in virtually all-distinguishable phenotypic features, such as cellular morphology, gene expression, metabolism, motility, and proliferative, immunogenic, angiogenic, and metastatic capacity [5,6,7]. A better understanding of tumor progression requires evaluating both the tumor cell-selective process and the interactions between biologically relevant tumor clones, and qualitative changes in the microenvironment from tissue to tumor. These mixed and dynamic parenchyma-stroma interactions result in tumor progression and heterogeneity.
Tumor development and progression can be regarded as a process of cell selection which, although assumed to occur randomly, has some factors like topography control the segregation of tumor cells within neoplasms[8,9,10]. The topographic intratumor heterogeneity suggests a differential selection of tumor cells, but can also be an expression of either selective clonal evolution or a simple passive byproduct of genetic instability [11]. The differential kinetic profile by topographic compartments has been related to lower cell turnover and apoptosis down-regulation in deep/peripheral compartments, resulting in accumulation of genetic alterations and segregation of tumor cells with differential genetic backgrounds as demonstrated in the adrenal gland, colon, and bladder. However, the coexistence of genetic alterations supports a key role in tumorigenesis, the topographic heterogeneity resulting from the accumulation of genetic damage. This concept is central and supports multiple sampling to assess the genetic abnormalities of neoplasms reliably. Numerous cell divisions are required for the emergence of full-blown malignancies and increased genetic instability, presenting plenty of opportunities for the appearance of various mutants. This genetic heterogeneity translates into phenotypic heterogeneity, and heritable phenotypes will, in turn, provide material for further selection forces. However, it is likely that a substantial fraction of phenotypic heterogeneity seen in tumors can arise from phenotypic plasticity and differentiation of cancer stem cells (CSC) and is therefore non-heritable. This process has also been linked with mismatch repair protein down-regulation, and it is unlikely to be related to hypoxia, which is more pronounced in central compartments. The role of cancer stem cells, hypoxia, and gene expression abnormalities are reviewed separately.
The heterotypic biology of neoplasms is an essential element to understand tumor growth. The underlying defect (clonal genetic alteration) may reside in stromal and not tumor cells, as reported in juvenile polyposis syndrome and ulcerative colitis hamartomatous polyps [12]. This finding suggests that at least initially the stromal cells are the neoplastic cells, which secreting factors drive the epithelial proliferation, and might thus eventually also be responsible for the induction of epithelial malignancy. This bystander role, mutations inducing stromal abnormalities that in turn induce epithelial neoplasia, has been called a landscaper effect: The microenvironment surrounding epithelial cells as a significant determinant of the disturbed epithelial architecture, differentiation, and proliferation (Figure 1).
Two principal mechanisms explain the impact of clonal heterogeneity on tumor evolution, modulating both progression and therapeutic escape: (a) Clonal diversity provides a more diverse genetic spectrum on which selection can take place; (b) The co-existence of genetically distinct clones within a tumor gives a network of biological interactions among distinct clones. As a consequence, the behavior of a tumor composed of distinct clones might be different from that of a monoclonal tumor or the sum of the individual clones [13,14].
Tumorigenesis is a dynamic selection process driven by genetic changes (mutations in broader terms) and abnormal gene expression [15]. In this context, higher genetic complexity is expected to provide more options for selection and a faster pace of evolution in heterogeneous neoplasms. As tumors grow, the cellular stress derived from replication results in mutations and expansion of sub-clones from the branching of the first precursor (monoclonal population). This selection results in an increased genetic complexity generated by branching from a tumor composed of multiple distinct clones [16]. Thus, clonally heterogeneous tumors can bring a more extensive variety of genetic variants to be tested by selection, which provides a more extensive adaptive landscape, increasing the probability of clones reaching fitness for challenges from several microenvironments (Figure 1).
The clonal composition of tumors can be especially important in determining responses to dramatic changes in the environment, such as changes induced by anti-cancer therapy. In this case, the pre-existence of resistant clones within a tumor can make the difference between tumor extinction (treatment success) and tumor evolutionary adaptation (treatment failure). A vivid illustration of the importance of clonal heterogeneity in therapeutic resistance can be found in malignancies from tyrosine-kinase receptor point mutations [17,18], or inactivating mutations of DNA repair mechanisms (resulting in genomic instability and highly sensitive to platinum compounds and poly ADP ribose polymerase inhibitors) [19]. The generality of the mutational mechanisms of acquiring therapeutic resistance remains an open question [20]. Nonetheless, intra-tumor heterogeneity is likely to represent a stiff challenge to therapeutic success, as more considerable genetic diversity within a tumor would be expected to increase the probability of the pre-existence of resistant cell types that could be selected for by treatment and ultimately result in a relapse of resistant tumors. Notably, in addition to the scenario of cancer therapy, selection for mutant variants rather than mutagenesis was proposed to be the key mechanism responsible for the carcinogenic action of a wide range of growth-limiting carcinogens [20,21,22].
Cancer morphologic and genetic heterogeneity is the expression of multistep tumorigenesis that leads to subclonal tumor cell populations with heritable traits (including primary, circulating and metastatic cells) and a microenvironment of ECM, fibroblasts, inflammatory cells and blood vessels. It is increasingly clear that understanding alterations within tumor cells are only part of the picture, and we need to understand interactions between tumors and their microenvironment to account for multiple aspects of tumor progression and therapeutic resilience [14,23,24]. Also, the differentiation hierarchy within tumors complicates the network, along with the importance of tumor microenvironment. A significant implication is the co-existence of phenotypically distinct clonal populations of tumor cells should inevitably lead to the formation of a network of biological interactions, which could be either direct or mediated by the tumor microenvironment. Some of the critical interactions that are likely to exist between distinct tumor clones are summarized below (Figure 2) [25]. Of note, most of the biological interactions are not mutually exclusive, and the net outcome for the interacting species will depend on the net sum of the different interactions. Inhibitory effects (competition, amensalism, antagonism) would result from progression interfering interactions, while stimulatory effects (commensalism, mutualism) are the consequence of progression boosting interactions (Figure 2). Therefore, it can lead to both augmentation and retardation of overall tumor growth and progression. Elucidation of biological interactions between populations of tumor cells might be a very formidable task; however, it can deepen our understanding of tumor biology and uncover new therapeutic targets. These interactions also determine the biologically relevant clones at the base of tumor dormancy (predominant arrested clones from cytostasis-differentiation), tumor growth (predominant proliferating clones from mitogenesis), invasion-metastasis (predominant invasive clones from motility), and therapy resistance (from cancer stem cells from viability) (Figure 1).
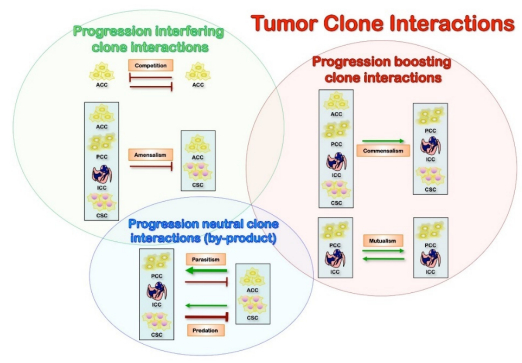 Figure 2. The increased genetic instability which leads to various mutations is caused by numerous cell divisions and the plasticity of the cancer stem cells. This clonal heterogeneity increases the probability of clones reaching fitness for challenges from several microenvironments. The landscaper effect is that the microenvironment surrounding epithelial cells is a significant determinant of the disturbed epithelial architecture, differentiation, and proliferation. Such examples of that are that the fundamental underlying clonal genetic alteration may reside in the stromal cells which may secrete driving factors for epithelial proliferation. Another factor is that chronic inflammation (Dendritic cells, TA macrophage, cytokines, and chemokines) promotes tumor onset and development through the production of myeloid-derived suppressor cells (MDSC) which inhibits both innate and adaptive immunity.
Figure 2. The increased genetic instability which leads to various mutations is caused by numerous cell divisions and the plasticity of the cancer stem cells. This clonal heterogeneity increases the probability of clones reaching fitness for challenges from several microenvironments. The landscaper effect is that the microenvironment surrounding epithelial cells is a significant determinant of the disturbed epithelial architecture, differentiation, and proliferation. Such examples of that are that the fundamental underlying clonal genetic alteration may reside in the stromal cells which may secrete driving factors for epithelial proliferation. Another factor is that chronic inflammation (Dendritic cells, TA macrophage, cytokines, and chemokines) promotes tumor onset and development through the production of myeloid-derived suppressor cells (MDSC) which inhibits both innate and adaptive immunity.Commensalism is a positive interaction in which one interacting party benefits the other without itself being affected. The tumor cell-stromal cell interaction in itself is a form of commensalism because it has been demonstrated that these nonmalignant cells support and even enable tumor growth [26]. An example of this type of interaction can be found in healthy tissues: Mammary epithelial cells from estrogen receptor null mice (ERα-/-) fail to grow and do not develop branching structures upon transplantation into cleared fat pads. However, when mixed with wild-type epithelial progenitors, ERα-/-cells do proliferate and contribute to different lineages and various parts of the mammary gland [27]. Also, nearby cells can protect each other from a set of host defenses that neither could survive alone. Cooperation can evolve as by-product mutualism among genetically different tumor cells [26]. A recent publication from the Weinberg laboratory demonstrated the relevance of this interaction in a human xenograft model, where an "instigator" tumor cell line augmented the proliferation and metastasis of genetically distinct "indolent" tumors that were incapable of forming macroscopic outgrowths [28,29]. In this case, the "instigation" was mediated by systemic effects, which could at least partially be attributed to the secretion of SPP1 (osteopontin) by the "instigator" cells [29].
Although not experimentally validated, this interaction can account for the co-existence of clonal variants that differ in their angiogenic potential. If the fitness benefit of angiogenic factor production is higher than the associated fitness cost, an angiogenic clone can undergo competitive expansion and reach a stable equilibrium with non-angiogenic "free rider" clones. Interestingly, mathematical modeling shows that, under certain physiologic conditions, "free riders" capable of faster proliferation can out-compete the angiogenic clone, leading to the collapse of the tumor [30]. This prediction can potentially explain spontaneous regression of neuroblastomas accompanied by massive necrosis [31].
Mutualism (cooperation) is a positive interaction in which both interacting parties can benefit from each other. Positive interactions between species have been widely documented in natural ecosystems. Mathematical modeling suggests that mutualistic interactions can lead to the coexistence of distinct species even under competitive contexts [26,32]. It has been suggested that mutualistic interactions between distinct tumor clones can play a significant role in tumor evolution by maintaining the survival and proliferation of tumor cells until one of the clones achieves a "full deck" of malignant mutations required for clonal dominance and full-blown malignancy [26,33]. To the best of our knowledge, however, such interactions between distinct clonal tumor populations have not been experimentally documented.
Competition is likely to be the most active and most significant biological interaction between tumor cells [34]. Mechanisms to limit the competitive outgrowth of mutant cells include natural tumor-suppressive mechanisms. Robust oncogene activation triggers tumor-suppressive networks, resulting in senescence or death of mutated cells [35]; or oncogenic mutations that can trigger stronger proliferation without engaging intracellular tumor suppressors resulting in loss of stemness (extrinsic tumor suppressor mechanisms) [36]. Moreover, the spatial organization of normal tissues can limit the role of competition between genetically distinct cells even further, confining stem cells to small pools [37,38]. However, carcinogenic and growth-limiting conditions can substantially modify the fitness landscape, allowing for the competitive outgrowth of ontogenetically mutated cells, thereby initiating malignant evolution [39,40]. Tumor progression is associated with further loss of tumor-suppressive mechanisms and disintegration of normal tissue morphology; thus, tumors start to resemble ecological systems rather than integrated tissues, and competition becomes the most robust biological interaction. The limited nature of resources further strengthens the role of competition: While, under tissue culture conditions, tumor cells are capable of endless exponential growth, clonal expansion within tumors is severely constrained by the limited availability of oxygen, nutrients, growth factors, and space (habitable niches). Limited resources intensify competitive interactions both within and between interacting cancer subclones. In large, spatially homogeneous populations, competitive interaction results in the fixation of a clone with the highest fitness value [25,37,38]. However, fixation can be inhibited by the spatial organization, which limits competition to within clones. Also, the existence of regions with different selective pressures can mediate the coexistence of clonally distinct populations. Finally, a stable coexistence of multiple clones is also possible when fitness is density-dependent [30].
Amensalism is an interaction in which one interacting party is inhibited by the other without being affected itself. Under the competitive context, this interaction is also referred to as "interference competition." As tumors grow under conditions of limited resources, amensalism interactions can provide a competitive advantage to a clone that can inhibit other clones while being (at least relatively) resistant. An example of this type of interaction between genetically distinct human cells can be found in Bcr-Abl-driven leukemias [41]. The existence of this kind of interaction has also been documented among distinct tumor clonal populations, both in cell culture and in vivo. The concept of amensalism can be extended to interactions between primary and metastatic tumors, as many human and experimental tumors can suppress metastatic outgrowth by inhibiting angiogenesis or inducing dormancy of single disseminated tumor cells through uncharacterized mechanisms [42]. Thus, amensalism can work as a weapon in competitive "warfare" between distinct clonal populations, which can lead to the stable coexistence of distinct clones in tumors.
Antagonism is an interaction in which one interacting party can capture biomass from the other one. This interaction is widespread in natural ecosystems, and there are clear parallels between organism-tumor and host-parasite interactions. However, antagonism is unlikely to be relevant for the interactions between distinct tumor clones.
Neoplasms are not static entities. They start from a genetically normal cell and conclude with billions of malignant cells that have accumulated numerous mutations during tumorigenesis, including the emergence of positively selected mutations ("drivers") and the accumulation of neutral variation ("passengers") [13,43]. Clonality is a key concept for our current understanding of tumor biology and comprises both clonal origin and expansions, which contribute to both tumor initiation and promotion [44,45,46,47]. Clonality tests cannot be interpreted in isolation; they will be meaningless without knowing the effect of a particular marker on cellular kinetics and the interrelationships of that marker with other genetic alterations that are present in a given neoplasm. This dynamic aspect is essential to get robust results and to avoid misinterpretations that might devalue the findings. As with many other issues in tumor biology, it cannot be based on single markers. A complementary approach that takes into consideration the technical limitations is essential to avoid the problems. Several markers have been used to assess tumor clonality [45,46,48], including X-chromosome inactivation, loss of heterozygosity (in particular targeting polymorphic regions of tumor suppressor genes), and mutation analysis. The existence of clonal heterogeneity has been documented for a variety of malignancies, but due to multiple technical challenges, the available data are mostly fragmentary, with the extent of clonal heterogeneity and the dependence of clonal heterogeneity on tumor type, subtype, and disease stage remaining mostly unexplored. It is useful to distinguish cellular genetic heterogeneity (differences at the level of single tumor cells) from clonal genetic heterogeneity (differences that have been amplified by clonal expansion) [49]. Focusing on clonal heterogeneity instead of cellular heterogeneity eliminates some of the "noise" of tumor evolution, as many of the variants detectable at the level of individual cells fail to clonally expand because of their occurrence in a cell that has lost stem cell properties, unfavorable effects on fitness, or simple stochastic reasons. However, "clonal heterogeneity" will not necessarily be completely "noise-free, " as clonal expansion does not necessarily prove the particular value of a mutation [50].
Neoplastic cells reveal genetic alterations that explain the acquisition of advantageous cell kinetics and invasion capacity (local and distant), most of them acquired. This constellation of alterations is most likely related to multiple cooperative genetic abnormalities that explain the biologic and clinical progression [1,2]. In this scenario, we need to consider that the first genetic alteration has not to be necessarily the irreversible abnormality leading to a clinically detectable neoplasm because genetic alterations can link to apoptosis or may be counterbalanced by other genetic changes resulting in no clinical growth. In inherited cancer syndromes, the first genetic alteration is known, but on its own does not explain clonal growth, the neoplastic lesion displaying additional alterations that correlate with the clinical presentation [45,46]. There are also genetic alterations such as fusion genes described in neoplasms and thought to be initiating an event that is also present in inflammatory conditions; in these circumstances, the evaluation will depend on the agreed definition of a given neoplasm [51]. The characteristic finding in all these scenarios is that knowing the first genetic event does not guarantee a clonal growth unless the additional collaborative alterations support an advantageous cell kinetic resulting in a neoplasm [8,9,47,52,53,54,55]. For these reasons, a robust clonality test must consider evaluating multiple markers that together can support or refuse a common progenitor for the lesion.
Neoplasms progress through a multistep process in which they acquire the genetic abnormalities, but the number of genetic alterations and the sequence of these alterations is not known [1,2]. It should also be noted that, from a perspective of selection operating in the evolution of tumors, stable, heritable changes in gene expression due to epigenetic alterations are indistinguishable from similar changes caused by alterations in DNA sequences. Silencing of gene expression by hypermethylation of promoter regions is frequently observed in cancers [56,57]; therefore, heritable epigenetic changes should be included in considerations of clonal evolution. In common neoplasms, we know the most frequent sequences of genetic abnormalities, but this is not going to be the critical pathway for all neoplasms in a particular location. Considering all these "uncertainties, " the most sensible approach will be the statistical one by testing the so-called null hypothesis. In clonality analyses, the null hypothesis to test will be if the samples are different, in other words, if they come from different progenitors. This assumption does not assure that the samples are identical or are derived from the same progenitor. The strength of the analysis will depend on the number of markers tested and the percentage of informative cases for each marker in a standard sample. The higher the values for these two parameters, the more reliable are the result obtained. Testing pathway-independent and dependent markers would be the most sensible approach for clonality assays, including those contributing to the acquired capabilities and processes already identified in neoplasms [1,2]. Partial approaches are valid, but they will not provide the same strength.
The main limitations would be the heterotypic biology of solid tumors and the biologic heterogeneity, as a byproduct of tumor progression, and the technical aspect of preventing artifacts [1,58,59,60]. The core mechanisms involved in tumor progression would require maintaining the cellular survival, and proliferation capacity for expansion, along with invasion and cytostasis-differentiation. These four principal mechanisms must cooperate in the same direction, and result from the interaction between tumor cells and the microenvironment, from which tumor-promoting and tumor-inhibiting signals can be observed; the balance of these signals will determine the evolution and, eventually, the progression of the neoplasm. The dynamic nature of neoplasms must be taken into account. There is a continuous selection process of tumor cells that contribute to the biologic progression and results in segregation of genetic abnormalities by conditions or topography [10,53,54,55,61]. Detailed sampling protocols to consider predictable heterogeneity (such as the topographic heterogeneity) and protocols including quality controls for proper steps during the tests are absolute requirements to have robust results [45,46,58]. These interactions are central elements for cellular selection, tumor progression, and intratumoral heterogeneity, which are best analyzed by stochastic models.
Different kinds of "precancerous" or "premalignant" lesions are often misused, including four biological types of lesions based on their fate. (Ⅰ) Those that progress to more advanced stages, including cancer; (Ⅱ) those that continue to grow without qualitative change; (Ⅲ) those persisting with no or minimal growth and no qualitative change; and (Ⅳ) those that may regress. Alternative pathways of progression from intraepithelial neoplasms must be considered to understand tumor natural history. Tumor heterogeneity and fully established genetic and kinetic features of malignancy in intraepithelial neoplasms and topographic compartments of invasive malignancies are key factors for this process. These two concepts are closely related to tumor initiation, have been developed for epithelial neoplasms and corroborate the idea of multistep tumorigenesis and accumulation of cooperative genetic abnormalities ("gatekeeper" and "caretaker" pathways) [4]. The paradigm is the idea of intraepithelial neoplasm/malignancy, being harder to extrapolate the idea to non-epithelial lesions. These lesions would be meaningful when they are present in structures with anatomical boundaries, and the cells do not recirculate/migrate in physiologic conditions, regardless of the lesion size. Regarding anatomical considerations, it is the basement membrane and not the tumor capsule the limiting structure.
The presence of a single genetic alteration cannot be considered diagnostic of malignancy even for early stages [1,62]. These problems preclude establishing reliable diagnoses of follicular carcinomas in-situ for encapsulated neoplasms carrying PAX8/PPARγ fusion genes, or lymphomas in-situ, even for lesions that initially carry molecular changes reported in malignancy. The opposite situation is equally essential: Histologically confirmed intraepithelial lesions are considered precursors, but they can accumulate genetic alterations and show cellular kinetic features of malignancies, as reported in MEN 2A [55,63].
Although the existence of intra-tumor phenotypic heterogeneity has been recognized from the early days of experimental cancer research, the relative contributions of heritable and non-heritable mechanisms are still not evident, and yet the nature of tumor heterogeneity can have profound implications both for tumor development and therapeutic outcomes. Tumor evolution has often been depicted as successions of initiations and promotions of mutated cells in clonal expansion rounds, where every new series is driven by the acquisition of additional kinetically-advantageous mutational events (selection process) [47]. This following process of stochastic acquisition of the leading mutations drives tumor progression, as a result of proliferation and increased genomic instability that produce growing selection [9,10,61,64]. Only minority of random mutations are selectively advantageous, while a significant fraction of mutations will be discarded by selection. Furthermore, many neutral or even mildly disadvantageous mutations can be retained in the population or even undergo some expansion due to genetic drift. Moreover, the long-term evolutionary success of mutations providing a decisive selective advantage is not granted. As a consequence, some of the mutations that are selectively advantageous at certain stages of tumor progression and can trigger substantial clonal expansion may lead to evolutionary dead ends and, therefore, may not be present in a fully malignant tumor. The complexity of tumor evolution is further influenced by the ongoing alterations of tumor microenvironment associated with tumor progression [14], which are likely to alter the selective pressures experienced by tumor cells. Therefore, at the microscopic level, tumor evolution is likely to be nonlinear, and substantial genetic heterogeneity is expected in tumor cell populations [10,53,61,63,65].
The microenvironment controls the survival and growth of healthy cells within defined environmental niches. Since normal cells lack necessary autonomous survival signals, they will not survive an inappropriate microenvironment [66]. Detachment-induced cell death (anoikis) is the proposed mechanism preventing normal cells from leaving their original environment and seeding at inappropriate locations [1]. Malignant cells start interacting with the surrounding ECM to evade local tissue control and avoid anoikis during tumor development and progression [67]. A bidirectional relationship is initiated between tumor cells and its surrounding stroma as the first step to invasive growth on metastatic spreading [68].
Classical multistage modeling of tumorigenesis evolves through the processes of local proliferative lesions (tumor initiation and promotion or selection), and acquisition of invasion-metastatic potential (tumor progression). Tumor promotion consists of the selective (clonal) expansion of altered cells to form focal lesions [44,69]. Within this definition, the process of promotion is mainly a quantitative phenomenon (many cells arising from a single cell), while no qualitative changes are necessarily implied. Human tumors arise from single cells that have accumulated the necessary number and types of heritable alterations. Each such cell leads to dysregulated growth and eventually the formation of a tumor. Despite their monoclonal origin, at the time of diagnosis most tumors show a surprising amount of intratumor heterogeneity in all measurable phenotypes; the evolutionary dynamics of heterogeneity arising during exponential expansion of a tumor cell population, in which heritable alterations confer random fitness changes to cells [70]. The loss of self-regulatory mechanisms during tumor progression results in increasing levels of tumor cell heterogeneity, and qualitative changes that generate distinct cellular subclones with different phenotypes [46,47]. Such a background represents the landscape for the full deployment of tumor progression.
These two unique processes, the mainly quantitative process of tumor promotion and the intrinsically qualitative process of tumor progression, are driven by two distinct microenvironments: The tissue and the tumor microenvironments [21,22,71]. The tissue microenvironment refers explicitly to the local environment surrounding altered cells during their selective clonal expansion to form focal proliferative lesions. Conversely, the tumor microenvironment describes the unique biological milieu that emerges inside focal proliferative lesions as a consequence of their altered growth pattern [21,22,71]. Such new biological niche is characterized by a tissue architecture which is not developmentally programmed and is bound to pose significant challenges for cell survival, due to altered/inadequate supply of oxygen and nutrients. These conditions, in turn, can lead to biochemical and metabolic alterations that can profoundly impact on the fate of the cell populations inside focal lesions [72].
The available evidence is consistent with the hypothesis that focal lesions result from the clonal expansion of altered cells; if so, how does a focal lesion develop from those rare altered cells? Theoretically, there are at least two different (and not mutually exclusive) possibilities:
(1) The altered/initiated cell is already endowed with some degree of inherent growth autonomy and starts to replicate unchecked, forming a focal lesion and then, after some further steps, a full-blown cancer [73]. The analysis of several multistage models of cancer induction has led to the conclusion that initiation per se does not result in any significant growth of preneoplastic and neoplastic lesions, and the appearance of the latter is heavily dependent on the presence of a promoting/selective environment [74]. Furthermore, transplantation experiments have convincingly demonstrated that different types of altered cells do not display any evidence of growth autonomy when transferred in a typical tissue environment of young animals in vivo [40]. By analogy, altered, putative initiated cells can be found in the skin of several healthy human subjects, suggesting that their presence per se not be necessarily associated with selective clonal growth, and additional (promoting/selective) influences must be enforced when the latter does occur [75]. Also, epidemiologic evidence on smoking and lung cancer suggests that clonal expansion of cells is much more relevant than new mutations [76].
(2) The other possibility is that the single altered cell does not express any significant degree of growth autonomy and is still under the control of standard homeostatic mechanisms; if this is the case, its selective clonal growth must be linked to the dynamics of cell turnover typical of the tissue where it resides. Thus, specific alterations of these dynamics could translate into a promoting effect on any putative altered cells present in that tissue. It is self-apparent that, within this perspective, the tissue microenvironment surrounding rare initiated/altered cells is given a central role in their selective emergence as focal proliferative lesions. In this context, the neoplastic development is a biologic process that is not directly caused by the inciting agent acting on a passive target but results from the interaction of living structures (cells, tissues, and organs) with those agents [77,78].
Rare initiated cells could withstand certain types of growth suppression imposed on surrounding healthy cells, thereby acquiring a proliferative advantage under appropriate "selective" conditions. Furthermore, the growth of such resistant cells to form focal proliferative lesions could be induced by physiological homeostatic stimuli, as it occurs during tissue regeneration and turnover, and these rare clones could emerge because the bulk of surrounding normal cells was unable to respond to those stimuli [79]. Incidentally, the work of Farber was the first to provide evidence that tumor promotion could be interpreted (and in fact defined) as a process of cell selection, thereby introducing a Darwinian perspective in the analysis of carcinogenesis [80]. Thus, early phases of cancer development, far from being exclusively cell-autonomous, appeared to be heavily dependent on environmental influences and in fact could be interpreted as adaptive reactions to altered conditions in the surrounding tissue [77]. Precisely, if the growth potential of normal cells in a given tissue was severely impaired, this could translate into a driving force for any altered/initiated cells to expand and compensate for the (relative) impairment of their surrounding counterparts. As a consequence, cancer development is a process to be considered and analyzed at tissue/organ level, not just at single cell level, and a role for the tissue microenvironment in this process is defined [12,77,81].
As the vast majority of carcinogens can both (Ⅰ) exert growth-suppression in the target tissue, possibly as a consequence of the inflicted DNA-damage [82], and (Ⅱ) induces rare altered/initiated cells with a resistant phenotype [83]. A clear outcome of such combined effects includes the possibility of a selective expansion of the resistant cell population [80,82]. As random mutations are more likely to damage the function of the genome rather than to improve it [80], the same genotoxic agent can both initiate the carcinogenic process and exert a promoting/selective effect on rare initiated/altered cells. These effects can are achieved by limiting the proliferative potential and imposing other cytotoxic effects in the bulk of the surrounding cells [84].
Cell transplantation experiments provide a direct testing of the hypothesis that a growth-constrained micro-environment can represent a dominant driving force during tumor promotion. Under growth-constraint endogenous conditions, transplanted altered cells could selectively expand in the recipient's liver, forming hepatocyte nodules and eventually progressing to hepatocellular carcinoma. Importantly, no nodular growth was observed when a similar preparation of hepatocytes was injected into usual, untreated recipients [40], suggesting that nodule cells had no inherent growth autonomy. The selective growth of altered cells in this system occurs under the influence of homeostatic mechanisms which are similar to, and possibly coincide with those controlling normal cell turnover [85].
One critical situation to be considered in this context is aging. Aging represents the significant risk factor for neoplastic disease (albeit it is not an avoidable risk) [86]. It is characterized, if not defined, by a generalized decrease in the functional proficiency of several organs and tissues, including a decline in their proliferative potential [87]. It has been shown that the microenvironment of the aged liver can support the growth of a transplanted epithelial cell line, while that of the young recipient is not [88], being able of stimulating the clonal expansion of transplanted normal hepatocytes [89]. This phenomenon does suggest that the liver microenvironment associated with aging is also clonogenic, and could, therefore, foster the emergence of altered cell populations. It appears reasonable to propose that such clonogenic potential might be linked, at least in part, to the growth-constrained environment associated with aging [89]. It is likely that stromal fibroblasts also play a role in such age-associated clonogenic effect. Senescent fibroblasts can, in fact, stimulate early growth of both grafted normal and tumor epithelial cells, suggesting that they can mediate, at least in part, the effect of aging on the parenchymal component of various tissues [90,91]. However, this effect can translate into selective growth of rare altered cells if the majority of surrounding counterparts are relatively impaired in their proliferative capacity, as it occurs during aging [92].
From a more general standing point, chronic tissue injury and inflammation lead to both impaired function and an increased risk of neoplastic disease in the target organ [93,94]. It is reasonable to consider that a common factor in all these instances could be the progressive exhaustion of the functional and proliferative capacity of parenchymal cells, paving the way for the selection of rare variant cells with an altered phenotype [95]. Consistent with this interpretation is the finding of diffuse or focal parenchymal atrophy concomitant with proliferative lesions of putative clonal origin [96,97,98,99,100]. The "proliferative inflammatory atrophy" of the prostate has been recognized as a risk factor for prostate carcinogenesis, and it is characterized by clusters of proliferating prostatic cells arising in areas of the atrophic epithelium [97], suggesting their possible regenerative significance [98].
In this context, defects in DNA repair pathways (a known cancer risk) result in increased probability of critical genetic alterations occurring in rare cells, and this will lead to the emergence of the neoplastic phenotype. However, such interpretation largely overlooks the consequences that the defective DNA repair might have on the bulk of the tissue and their possible contribution to the increased carcinogenic risk that is seen in these conditions. Defects in DNA repair pathways can be associated with accumulation of widespread DNA damage [101]. It is reasonable to assume that such randomly inflicted damage will impair genome function [101], rather than improve it; in fact, accelerated aging has also been associated with altered DNA repair capacity [102]. Thus, the above considerations suggest that carcinogenesis related to defective DNA repair is also interpretable as the result of at least two primary biological components: (Ⅰ) Induction of rare altered cells and (Ⅱ) selection of such cells within a functionally impaired tissue environment.
The clonal amplification of altered cells fuels carcinogenic process by increasing the likelihood that further genetic changes will occur in those dividing cells, towards the acquisition of a fully malignant phenotype [103]. According to this view, tumor promotion consists primarily, if not exclusively, in the clonal amplification of altered cells, which is per se sufficient to increase the risk for additional genetic hits, thereby fostering cancer development. While such interpretation might be theoretically appealing, it is pertinent to point out that early focal lesions resulting from tumor promotion (i.e., polyps, papillomas, nodules) are not associated with the emergence of cellular sub-clones, as the hypothesis above would predict. It is also worth reiterating that promotion entails a mainly quantitative phenomenon of selective amplification and appears to be driven in many cases by physiological mechanisms involved in normal tissue turnover and reaction to injury [104]. In this context, the term focal lesion refers to a discrete collection of cells displaying a growth pattern and histological appearance, which are sufficiently distinct from that of the surrounding tissue resulting from the selective expansion of initiated/altered cells. According to this view, the focal nature of early lesions represents the critical endpoint of tumor promotion in that it forms the basis for the establishment of a new biological niche, with a fundamentally altered tissue architecture, which is referred to as the tumor microenvironment [71,105]. The emergence of the tumor microenvironment from the clonal growth of altered cells represents indeed the quantum change brought about by the tumor promotion phase in the natural history of cancer development. There is a crucial role of the tumor microenvironment in controlling tumor progression and metastasis. However, the formation of a specific environment supporting CSC/CIC survival and growth (the CSC niche), its anatomical organization and the cellular and molecular mediators of these effects, are still under investigation [106]. Brain tumor CSC/CIC are localized in a vascular niche that is supposed to provide factors promoting their self-renewal [107,108]. Perturbation of this niche results in a compromised ability CSC/CIC to promote tumor growth within the brain [109,110]. These observations demonstrate the relevance of the vascular niche to tumor pathophysiology and suggest the possibility to target the niche itself, and associated molecular events, to impinge on CSCs for therapeutic purposes [111,112].
Many signaling cascades controlling cell growth and proliferation are deregulated in cancers, resulting in excessive activation of downstream pathways, by signaling pathways that are not functional under nonpathological conditions, and by alterations in gene expression patterns [1]. These situations may involve newly formed interactions between microenvironmental factors and tumor cells and between different microenvironmental factors [23,113,114,115,116]. These interactions constitute a fertile ground for the establishment of circular chains of tumor progression-enhancing events described as self-perpetuating cycles. Monocyte chemoattractants CCL5 and CCL2 secreted by breast tumor cells may induce monocyte infiltration to the microenvironment of breast tumors. The resulting tumor-associated macrophages may secrete TNFα, which induces or upregulates the secretion of several pro-malignancy factors from the tumor cells such as matrix metalloproteinases. TNFα also further upregulates the secretion of CCL5 and CCL2, which drive the merry-go-round for another cycle [117]. A recent study indicated that a vicious cycle involving the CXCR3–CXCL10 axis and IFNγ works in colorectal carcinoma progression [118]. CXCL10 secreted from CXCR3-expressing colorectal carcinoma cells promotes, by an autocrine mechanism, some progression-promoting functions in these tumor cells. CXCL10, at the same time, attracts CXCR3-expressing Th1 cells to the tumor site. The infiltrating Th1 cells secrete IFNγ, which, in addition to its immune functions, promotes the release of CXCL10 from IFNγ receptor-expressing colorectal carcinoma cells while up-regulating CXCR3 expression. This expression further enhances the capacity of the colorectal carcinoma cells to respond to CXCL10-mediated pro-malignancy functions. Self-perpetuating cycles involve multiple participants, each of which may serve as a target for specific adjuvant treatment of metastasis. However, cancer cells are endowed with the capacity to bypass regulatory roadblocks or pathways by a variety of mechanisms [119,120]. Every self-perpetuating cycle described above (and most others) involves numerous cellular interactions. Because the blocking of each interaction may be subject to a bypass strategy, we need to use multiple therapy modalities targeting most, if not all, components of such self-perpetuating cycles. A series of biochemical and metabolic changes are typically associated with the tumor microenvironment, is attributable, at least in part, to the altered blood and nutrient supply [72]. These changes can both induce and select for cell variants within the original focal cell population, setting the stage for the emergence and evolution of cell clones which represent the biological hallmarks of tumor progression [72,105,121,122,123]. Consistent with this interpretation, the analysis of multistage models of carcinogenesis has documented that the extended phase of tumor progression, leading from discrete focal lesions to the overt neoplastic phenotype, is a self-perpetuating process and does not depend on external manipulation, as it is the case for tumor promotion [104]. Thus, the unique microenvironment inside focal lesions, with its associated biochemical and metabolic alterations [72,105,123], appears to be sufficient to drive tumor progression.
Chronic inflammation promotes tumor onset and development through nonimmune and immune mechanisms. The nonimmune mechanisms include three essential aspects. (Ⅰ) The production of reactive oxygen species (ROS) such as peroxynitrites, which cause DNA mutations that contribute to genetic instability and the proliferation of malignant cells [124]. (Ⅱ) The production of proangiogenic factors such as vascular endothelial growth factor (VEGF), which promote tumor neovascularization [125]. (Ⅲ) The production of matrix metalloproteases, which facilitate invasion and metastasis [126]. The efficient immune mechanism is the perturbation of myelopoiesis and hemopoiesis, which causes a deficiency in Ag-presenting dendritic cells (DC) and dysfunctional cell-mediated antitumor immunity [127]. A primary culprit in this latter deficiency is the production of myeloid-derived suppressor cells (MDSC), a population of immature myeloid cells that are present in most cancer patients and mice with transplanted or spontaneous tumors, and inhibits both innate and adaptive immunity. They are likely to subvert immune surveillance and prevent an individual's immune system from eliminating newly transformed cells. In people with established cancer, they are likely to be a significant factor in preventing the efficacy of immunotherapies, such as cancer vaccines, that require an immunocompetent host [128,129]. MDSC cause immune suppression in most cancer patients, where they impede all immunotherapies that require an active immune response by the host. They may also facilitate the transformation of premalignant cells and promote tumor growth and metastasis by suppressing innate and adaptive immune surveillance that would otherwise eliminate abnormal cells. The induction of MDSC by proinflammatory factors identifies the immune system as another contributing mechanism by which chronic inflammation contributes to the onset and progression of cancer.
In the tumor microenvironment, tumor-associated macrophages (TAMs) constitute the majority of tumor-infiltrating leukocytes. MDSC have been identified in most patients and experimental mice with tumors based on their ability to suppress T cell activation. Two distinctive TAM subpopulations have been defined. Classical, or M1, macrophages are characterized by the expression of high amounts of iNOS and tumor necrosis factor-α (TNF-α), whereas alternatively activated M2 macrophages typically produce ARG1 and IL-10 [130,131]. At the tumor site in wild-type mice, TAMs are predominantly M2-like macrophages, which are the cells primarily responsible for suppressing T cell-mediated antitumor responses and promoting tumor progression, metastasis, and angiogenesis [132,133,134]. M1 macrophages, in contrast, exhibit, a tumoricidal effect [135,136,137]. Monocytic MDSCs and TAMs share several characteristics, such as expression of the monocyte and macrophage markers F4/80 and CD115, as well as inducible expression of iNOS and ARG1 [131,132,138]. Accumulating evidence suggests that, upon entering tumor tissues, MDSCs may differentiate into TAMs, leading to elevated IL-10 production, inhibition of T cell responses, and promotion of angiogenesis [139]. However, the mechanism behind regulation of MDSC differentiation remains unclear [131,139,140].
Macrophages within the tumor microenvironment facilitate angiogenesis and extracellular matrix breakdown and remodeling and promote tumor cell motility. There is a direct communication between macrophages and tumor cells that lead to invasion and egress of tumor cells into the blood vessels (intravasation). Thus, macrophages are at the center of the invasion microenvironment [141]. Mononuclear phagocytes are recruited in large numbers as primary monocytes from the circulation to diseased tissues, where they accumulate within ischemic/hypoxic sites terminally differentiating into inflammatory and tumor-associated macrophages or myeloid dendritic cells (DCs). The cytokine MIF is overexpressed in tumors and is associated with tumor proliferation, angiogenesis, and metastasis. Hypoxia, a hallmark feature of tumors, increases MIF expression from tumor cells. Inhibition of transcription and translation significantly decreased MIF production, suggesting that hypoxia-induced secretion of MIF is via an alternative pathway [142]. Hypoxia-mediated changes in mononuclear phagocyte gene expression and functional properties under different pathologic situations demonstrate that oxygen availability is a critical regulator of their functional behavior. Experimental evidence is showing that hypoxia modulates in primary monocytes the expression of a selected cluster of chemokine genes with a characteristic dichotomy resulting in the up-regulation of those active on neutrophils and the inhibition of those predominantly active on monocytes, macrophages, T lymphocytes, NK cells, basophils and DCs is reported. A negative regulatory role of hypoxia on monocyte migration is exerted through several alternative or complementary mechanisms and results in monocyte "trapping" within ischemic/hypoxic sites of diseased tissues. Hypoxia differentially regulates immature DCs (iDCs) by expression down-regulation of chemokines and upregulation of chemokine receptors, thus promoting the switch from a proinflammatory to a migratory phenotype of iDCs by, respectively, reducing their capacity to recruit other inflammatory leukocytes and increasing their sensitivity to chemoattractants [143]. A partial overlap exists among mononuclear phagocytes at various differentiation stages in the expression of a cluster of hypoxia-responsive genes coding for regulators of angiogenesis, proinflammatory cytokines/receptors, and inflammatory mediators and implicated in tissue neovascularization and cell activation. Transcription pathways underlying hypoxia-regulated gene expression in monocytic lineage cells support a major role for the Hypoxia-inducible factor-1 (HIF-1)/hypoxia responsive element (HRE) pathway in monocyte extravasation and migration to hypoxic sites and in the activation of monocyte/macrophage proinflammatory and immunoregulatory responses by hypoxia both in vitro and in vivo. Additional transcription factors contribute to this effect, such as nuclear factor-kappa B (NF-kappaB), Ets-1, CCAAT/enhancer binding protein-alpha/beta (C/EBPalpha/beta), activator-protein-1 (AP-1), and early growth response-1 (Egr-1). Hypoxic transcription regulation of these transcription factors in primary human monocytes, and differentiated macrophages indicate the existence of both a positive and a negative O(2)-driven HIF-1-dependent feedback regulatory mechanisms [143].
Whether tumor-induced MDSC is normal cells halted in the intermediate stages of differentiation or whether they have diverged from the usual myeloid differentiation pathway and accumulated mutations is unclear. Direct comparisons of in vitro suppressive activity of splenic Gr1+CD11b+ cells from tumor-free mice vs. tumor-bearing mice are not consistent. MDSC that are mononuclear is considered "monocytic" and typically are CD11b+Ly6G+/–Ly6Chigh, whereas those with multi-lobed nuclei are "granulocytic/neutrophil-like" and have a CD11b+Ly6G+Ly6Clow phenotype [144,145]. MDSC also vary in their content of immunosuppressive substances, with different populations containing arginase [146,147], inducible NO synthase [148], and additional ROS [146,148]. In cancer patients, MDSC is typically CD11b+CD33+CD34+CD14–HLA-DR– and can vary in their expression of CD15 and other markers [144,145]. The variation in MDSC phenotype is consistent with the concept that MDSC is a diverse family of cells that are in various intermediate stages of myeloid cell differentiation. Tumor-secreted factors drive MDSC, and different tumors secrete different combinations of molecules. Therefore, MDSC phenotype will depend on the specific combination of factors within the tumor host. Because the myeloid population contains many different cell types and myeloid cell differentiation is a continuum of processes, MDSC may display several phenotypic markers that reflect the spectrum of immature to mature myeloid cells. This heterogeneity suggests that there may be no single marker or combination of phenotypic markers that precisely defines MDSC and that suppressive activity is the ultimate defining characteristic. It is also likely that, as this population of cells is further studied, additional subpopulations and markers will be identified. The heterogeneity of MDSC also complicates finding a single strategy for eliminating the cells. Pathologically distinct tumors produce different arrays and quantities of proinflammatory factors that induce MDSC. As a result, there is phenotypic heterogeneity between MDSC induced by histologically distinct tumors. There is also phenotypic heterogeneity within the MDSC population induced within a single individual. This heterogeneity may require identifying and then specifically target the crucial proinflammatory mediator(s) for individual patients or the specific type of tumor.
MDSC accumulation and activation are driven by multiple factors, many of which are identified with chronic inflammation. Early studies demonstrated that the inflammation-associated molecules VEGF and GM-CSF were associated with the accumulation of MDSC [149], suggesting that inflammation might facilitate immune suppression [150]. However, it was not until the proinflammatory cytokines IL-1β [151], and IL-6 and the bioactive lipid PGE2 were shown to induce MDSC that the significance of the association with inflammation was appreciated [147,152]. These later studies suggested that another mechanism by which inflammation promotes tumor progression is through the induction of MDSC that block immune surveillance and antitumor immunity, thereby removing barriers that could eliminate premalignant and malignant cells. MDSC are induced and activated by multiple proinflammatory mediators. MDSC accumulate in the blood, bone marrow, lymph nodes, and at tumor sites in response to pro-inflammatory molecules produced by tumor cells or by host cells in the tumor microenvironment. These factors include PGE2, IL-1β, IL-6, VEGF, S100A8/A9 proteins, and the complement component C5a.
The tumor microenvironment generates an active process that causes dynamic cellular selection. Hence it is selectogenic. Given that altered cells can be selected in a tissue microenvironment which is otherwise growth-inhibitory to surrounding counterparts, a relevant question pertains to the biochemical and molecular basis of such phenotypic resistance. Blagosklonny has proposed the existence of two broad types of resistance [20]. (Ⅰ) Non-oncogenic resistance relate to changes in drug metabolism and uptake, such that the rare altered cell can withstand toxicity compared to the rest of the population in that tissue. Such phenotypic resistance would still translate into the clonal growth of that rare cell, but no increased risk of neoplastic disease would be implied [20]. (Ⅱ) The oncogenic resistance is linked to the inability of the cell to sense or repair DNA damage and to activate effector mechanisms leading to cell cycle arrest and cell death. As a result, the affected cell is susceptible to acquire a "mutator phenotype, " i.e., the tendency to undergo a cascade of further mutations [1,8,80,153].
The mutator phenotype has been linked to a defect in mismatch repair (MMR) genes so that a cascade of mutations occurs in cancer-related genes. To justify the onset of a mutator phenotype in "sporadic cancers" (which are in fact the vast majority) we have to revisit some theories of carcinogenesis and their evidence base [10,64]. In sporadic cancers, the origin of the mutator phenotype has been attributed to chance, or to mutagens that selectively affect specific genes similar to MMR genes, or to a combination of the two. However, MMR is mutated only in a minority of cases, for example, colon cancers. The proportion of colon cancer with microsatellite instability (MIN) is small compared to cancers characterized by chromosome instability (CIN), whose onset has not yet been attributed to the failure of any specific gene repair such as MMR [8,153]. To explain the most common type of lesions that are found in non-hereditary cancers, chromosome aberrations, and CIN, we have to explain how the mutator phenotype originates. Also, a fundamental concept that has also emerged recently is that mutations–or instability–themselves are irrelevant if there is not a microenvironmental change that selects the cells carrying such mutations. Therefore, we will discuss first some examples of such a "selectogenic" microenvironment.
Chromosome instability (CIN) and microsatellite instability (MSI) have been described as two alternative pathways to cancer [1,8]. Chromosomal instability is defined as the ability of a cell to gain and lose chromosomes and is a feature of many types of cancer. Conversely, microsatellite instability is related to a defect in the DNA mismatch repair machinery (MSI cancers).
The net result of CIN is the deregulation of chromosome number (aneuploidy) and an enhanced rate of loss of heterozygosity, which is an essential mechanism of inactivation of tumor suppressor genes. Cytogenetic studies of bladder, lung, and colon tumors have shown that karyotype complexity, cell ploidy, and the number of structural changes found is closely associated with tumor grade and stage. It has been suggested that different environmental carcinogens can induce specific forms of genetic instability [154]. The available data demonstrate that exposure to specific carcinogens can indeed select for tumor cells with distinct forms of genetic instability and vice versa. These data offer potential clues to one of the remaining unsolved problems in cancer research, the relationship between environmental factors and the genetic abnormalities that affect tumorigenesis.
An understanding of cancer progression is incomplete without an analysis of metastatic tumors and disseminated tumor cells. Cancer is a systemic disease: malignant tumors shed a significant number of cells into the blood and lymph vessels, some of them developing in distant sites into metastases. Moreover, distant metastasis is responsible for the majority of cancer-related deaths, and, therefore, understanding the underlying biological mechanisms of it is of primary importance. The invasion/metastasis capability is closely related to cell motility and requires the cytoskeleton as a critical component, which is also essential during mitoses. The histological malignancy criteria frequently mirror the phenotype of actively proliferating cells and, less commonly, the phenotype of the invasive component at the periphery of solid organ neoplasms and deep compartment of luminal organ tumors. The last compartment shows lower cellular turnover and a higher incidence of genetic abnormalities [8,9,52,53,64]. These factors need attention when planning the evaluation of intratumoral heterogeneity and tumor progression, in particular, a combined evaluation of dynamic and genetic features to assess a selective process with profound redundancy and pleiotropism [1,47,155]. These biological foundations will enable a better therapeutic design, using the heterogeneity to improve patient's management.
There are two primary models to explain the intratumoral heterogeneity: A random process that would result in a patternless distribution or selection process that may be topographically linked and lead to identifiable compartments including intraepithelial components. The malignancy-associated genetic instability would determine an independent evolution in different tumor areas, regardless of the location (intraepithelial or invasive). Some so-called precancerous lesions show the genetic and kinetic features of established malignancies, including clonal proliferation with an accumulation of cooperative genetic abnormalities, and advantageous proliferation/apoptosis dysbalance [47,55,63,156,157]. High-grade lesions are the most reliably diagnosed intraepithelial malignancies and progress through multiple morphological and molecular steps, which can and most likely include the acquisition of invasion/metastasis capability. As the order of these changes is not pre-established, they can be present well before the morphological evidence of malignancy [158], which reflect intratumoral heterogeneity and explain the presence of metastasis in cytologically low-grade lesions (multiple parallel pathways) that not necessarily progress through the high-grade stage (direct pathways).
The connection between early neoplasm stages and metastasis is tumor heterogeneity and progression. Progression refers to the acquired capability of cell growth in surrounding or distant tissues, reflecting the acquisition of invasive capacities for intraepithelial lesions and metastatic capacities for invasive neoplasms. This ability reflects the interaction between tumor cells and the microenvironment. It would not be surprising to find the similar control for the tumor cell capabilities in both steps (stromal invasion and metastasis) [1]. This progression would be related to the accumulation of genetic abnormalities and selective segregation of tumor cells with invasive capabilities, which are frequently topographically distributed [9,52,53,159]. Clinicopathological studies have demonstrated lymph node metastases associated with histologically intraepithelial malignancies (i.e., breast, skin) [160,161]. Several intraepithelial foci showed more alterations than matched invasive foci, suggesting a more extensive genetic evolution for the former and supporting multifocality and independent clonal evolution of these coexistent carcinomas. The accumulation of genetic abnormalities in intraepithelial carcinomas is consistent with an advanced molecular stage and progression, along with topographic genetic heterogeneity [53,55,61,63,65,156]. Cancer cells can survive and proliferate only at specific secondary sites where there is an ideal environment that releases molecular mediators suitable for that type of cancer cells, still represents a main conceptual model of metastasis in modern cancer research [7,162,163,164]. Metastasis formation itself is a multi-step process that requires tumor cells to escape from the primary site, intravasate into the vascular or lymphatic circulation, migrate and extravasate into secondary organs [162,165]. Recent work has shed new light on the genetic, molecular and cellular basis of metastasis [166,167,168]. CSC/CIC might represent the unique subpopulation of cells with the potential to successfully form metastasis in a distant organ [169,170]. Metastasis formation, however, is a rather inefficient process, mostly due to the need for cancer cells to find a proper microenvironment for initiating tumor growth in a secondary organ. It has been proposed that to form metastases; primary tumors might produce factors that induce the formation of a suitable and appropriate environment in the organ where metastasis will be seeded. This suitable environment has to lead to the concept of the premetastatic niche, whereby a unique, permissive microenvironment in secondary target organs is induced over distance by the primary tumor [171,172]. In this sense, there is a striking parallel between CSC/CIC maintenance and expansion in the primary tumor and formation of metastases in a distant organ. Both events require a particular niche or microenvironment and might share many cues promoting self-renewal ability, migration and invasion, resistance to apoptosis, and increased resistance to cytotoxic drugs. The first in vivo experimental evidence that metastatic seeding requires the formation of a niche was the discovery that VEGFR-1+ BMDC colonizes target organs to form tumor-specific premetastatic sites, before the arrival of the metastatic tumor cells themselves [173]. In these locations BMDC expresses several hematopoietic markers, such as CD34, CD116, c-kit, Sca-1, as well as integrins (e.g., α4β1), chemokines and chemokine receptors (e.g., CXCL12/CXCR4), promoting either their homing to the target tissue or recruitment and attachment of metastatic tumor cells or both.
According to a traditional model of tumor development, tissue constraints constitute a major evolutionary bottle-neck in cancer evolution. The acquisition of metastatic ability is considered to be the final step in tumor development, contingent on the elaboration of all of the other hallmarks of cancer [1,2,3]. This model implies that metastatic tumors should be genetically similar to the bulk of primary tumor cells.
Many studies that compared the genetic composition of primary tumors and secondary metastatic sites have found very close clonal relationships between the two in the majority of cases [174,175,176]. Similarly, analysis of gene expression profiles revealed very similar patterns between primary tumors and metastatic sites, a scenario highly unlikely for genetically different clones [177,178,179]. Another prediction from the linear model of tumor progression is that different metastases should display close clonal relationships among each other. Indeed, this prediction is supported by a recent study that compared the genetic composition of anatomically distinct metastatic lesions in 29 prostate cancer patients using SNP arrays and CGH [180]. In all cases, different metastatic lesions within the same patients demonstrated close clonal relationships, signifying monoclonal origin [180]. This demonstration of monoclonality of metastatic cancers is especially impressive given that primary prostate cancers are frequently multifocal [181], and show substantial intra-tumor genetic heterogeneity [181,182].
While the evidence of the close genetic relationship between primary and metastatic tumors is compelling, some cases display dramatic divergence, challenging the model where the acquisition of metastasis is considered to be the last step of tumor progression. Radically different patterns of allelic losses, indicative of a high degree of genetic divergence, have been reported in primary tumors and lymph node metastases in prostate cancers [175], and between primary tumors and asynchronous metastases in breast cancers [176]. Highly divergent clonal evolution was also evident in a subset of cases in CGH studies of primary tumors versus lymph node metastases in breast cancers and of primary tumors versus metastatic tumors in renal cell carcinomas [174]. A recent report, comparing sequences of primary tumors and metastases in lobular breast cancers, revealed multiple mutations present only in metastases and several other mutations with increased frequency in metastatic sites [183]. Some of these genetic changes result in higher incidence of apoptosis of tumor cells of dormant metastases (more than three-fold higher) [184]. These data show that metastases remain dormant when tumor cell proliferation is balanced by an equivalent rate of cell death and suggest that angiogenesis inhibitors control metastatic growth by indirectly increasing apoptosis in tumor cells.
Vital clues about the clonal evolution of tumors and the relationship between primary tumors and metastatic sites can be gained by the analysis of disseminated tumor cells. Epithelial-specific markers, cytokeratins and epithelial cell adhesion molecule (EpCAM), can identify disseminated epithelial tumors in peripheral blood, bone marrow, and lymph nodes [185,186,187,188,189]. Klein and co-authors, using a PCR-based whole-genome amplification technique unexpectedly reported single disseminated tumor cells [190]. The authors demonstrated that disseminated tumor cells could be detected both in the bone marrow of patients and in mouse models of cancer at very early, pre-invasive stages of breast cancer development [158]. More support for the rapid dissemination of cancer cells came from CGH analysis of disseminated tumor cells from patients without overt metastases. These cells contained surprisingly few chromosomal aberrations [191], while higher-resolution LOH analysis did confirm the neoplastic origin of these cells and pointed to early events in tumor progression [192]. Impressive results were obtained for esophageal cancers, one of the most aggressive human carcinomas, where removal of primary tumors does not improve the chances of patient survival [193], suggesting the importance of relatively early metastatic spread. Lymph node and bone marrow metastases showed substantial divergence from primary tumors and among each other, suggesting parallel evolution shaped by microenvironment-specific selection forces. Interestingly, HER2 amplification in disseminated tumor cells was found to signify poor prognosis, whereas HER2 amplification in primary tumors had no prognostic value, further underlying the significance of divergent evolution between primary tumors and metastatic sites [194]. It should be noted, however, that since this analysis, based on the amplification of single-cell genomes, is associated with many technical caveats, critical interpretation of the data is highly warranted.
The obvious caveat of the studies of disseminated tumor cells is that the potential of any given disseminated single tumor cell to initiate a metastatic tumor is unknown. The mere fact that tumor cells survive at distant sites tells little about their ability to start a secondary tumor. Indeed, even normal epithelial cells were capable of extended survival in heterotopic locations in an animal model [195], and fetal cells are known to survive decades in women following pregnancy [196]. Also, most carcinomas display pronounced chromosomal instability, and, therefore, many disseminated cells might be a manifestation of "cell-to-cell heterogeneity, " i.e., they are products of genomic instability without any substantial chance of clonal expansion. Testing the relevance of these disseminated cells to subsequent metastasis formation is not trivial. The most compelling evidence comes from a mouse model of breast cancer where the growth of lung metastases paralleled that of the primary site from early points of tumor progression, and excision of mammary glands when primary tumors were still at the in situ stages did not prevent or delay the development of metastases [158]. Tumor cells that disseminated from early-stage tumors into the bone marrow were capable of lethal proliferation upon transplantation into lethally irradiated mouse recipients [158]. While these findings are compelling, their relevance to human cancer is not clear, especially considering that the genetic heterogeneity of disseminated cancer cells in mouse tumors appears to be less pronounced than that in human tumors.
Another group demonstrated that bone marrow aspirates from carcinoma patients contain disseminated tumor cells that are capable of proliferation in cell culture and that the extent of this proliferation is inversely correlated with patient survival [197]. CGH analysis of disseminated tumor cells from breast cancer patients confirmed their high divergence from primary tumors [198]. This study demonstrated substantially higher numbers of genomic abnormalities in disseminated tumor cells compared to the studies of Klein et al., despite similar staging of the tumors analyzed, suggesting that the ability of disseminated cells to form metastases is contingent on the acquisition of a vast number of genetic alterations. Additional evidence for parallel clonal evolution comes from the detection of metastatic tumors with the unknown primary origin, which constitutes a significant fraction of clinical cases [199]. Also, analysis of the growth kinetics of primary and metastatic tumors is inconsistent with the metastatic spread being a late event [200].
Currently, the majority of therapeutic decisions are being made based on the analysis of primary tumor specimens. This approach could only be justified in cases where the genetic compositions of primary and metastatic tumors are similar, especially when making decisions about emerging therapeutic approaches that target specific genetic changes. Also, elucidating the issue of early versus late origin of metastasis-initiating cells is essential for determining the usefulness of therapeutic approaches that target tumor cell invasion. The question of clonal heterogeneity within metastases remains unexplored, and, notably, clonal heterogeneity within primary tumors has limited direct therapeutic relevance, as in most cases primary tumors can be successfully removed by surgery, but this is most often not the case for metastatic outgrowths. Therefore, elimination of metastatic clones has to rely on adjuvant therapies, and it is likely that the success of these therapies will depend on the extent of the heterogeneity of these tumor cell populations.
Tumors arise from a process of replication, mutation, and selection through which a single cell acquires driver mutations which provide a fitness advantage by enhanced replication or resistance to apoptosis [1,2,201]. Any cancer progresses through a relatively narrow evolutionary trajectory [202], "jackpot" mutations, which endow mutant clones with the ability to achieve selective sweeps, are likely to be very rare [1,44,46,47]. Most of the spontaneous mutations that occur are neutral or deleterious, and, while grossly disadvantageous mutations will be discarded, neutral or near-neutral mutations can undergo expansions due to genetic drift and can sometimes reach fixation in small tumors [25]. The complexity of cancer progression can be understood as the result of multiple sequential mutations, each of which has a relatively small but positive effect on net cell growth. Tumor initiation and progression are dependent on rare, stochastic mutational events, and the chances of any given individual getting diagnosed with particular cancer within a limited timeframe are relatively low, suggesting that cancers have to be clonal in origin, i.e., they are likely to arise from a common progenitor. This consideration, supported by substantial experimental evidence, has led to a broad acceptance of the clonal origin of cancers [203,204], although there might be some rare exceptions in cases of hereditary types of cancer [205]. Rare mutations confer competitive advantage through cellular selection, resulting in clonal dominance. Even though most cancers start from a single mutated cell, mechanisms that constrain clonal evolution in populations of normal progenitor cells are no longer fully functional in tumors, allowing for an ongoing evolutionary process in populations of tumor cells [206]. Multiple rounds of proliferation, often counter-balanced by cell death, are required to produce visible tumors, and genomic instability, observed in most cancers, is expected to continually produce new mutations, which serve as raw material on which tumor evolution can work.
Genetic mutations can arise either due to errors during DNA replication or from exposure to genotoxic agents [1,2]. Therefore, a deterministic model of evolutionary dynamics will significantly underestimate the time to cancer. The closer approximation exploits the standard behavior of the system of propagating a Gaussian distribution of error types and takes into account stochastic effects in determining the speed of this traveling wave. Thus, stochastic effects can play a significant role even in vast populations. The number and type of mutations observed is the result of the size of the population at risk, the mutation rate, and the microenvironment of the evolving clone. The individual mutation rate can vary significantly due to genetic and environmental effects [207,208]. The model of cancer progression might add to the debate whether selection or mutation is the dominant force in tumor development [209,210,211,212]. The final course to malignancy will be determined by multiple mutations, each with a small and distinct fitness advantage, and these mutations occur stochastically.
Moreover, spatial constraints that exist within a tumor can slow down the achievement of clonal dominance by substantially inhibiting competition among different tumor cell clones [9,10,52,53,65,155,213]. The importance of spatial constraints for the emergence and maintenance of clonal heterogeneity is supported by experimental observations in colon cancer as well as in silico modeling [214]. In addition to providing barriers to competition, spatial intra-tumor heterogeneity can support the co-existence of genetically distinct clones by affecting selective pressures. Most tumors represent at least partially structured habitats where individual cells within a tumor are experiencing differences in interactions with ECM, physical contact with other cells (both tumor and non-tumor), and gradients of oxygen, nutritional and growth factors, and metabolites. Therefore, tumor cells within different parts of tumors could be expected to experience different selective pressures, leading to the selection of the various sets of mutations. Mathematical modeling suggests that, even in the absence of substantial spatial heterogeneity, evolving pre-malignant tumors are likely to be characterized by quasi-steady states with the co-existence of multiple populations with different "strategies". However, a clone that evolves toward invasive cancer will be capable of invading and destroying pre-malignant populations [215,216]. Every cancer will thereby be dependent on a unique complement of mutations that will determine its propensity to invade, its ability to metastasize, and its resistance to therapies. If this model is correct, then biological heterogeneity is a direct consequence of the tumorigenic process itself.
Hypoxia or oxygen deficiency is a salient feature of locally advanced solid tumors resulting from an imbalance between oxygen (O2) supply and consumption. Heterogeneities in tumor blood flow are associated with cyclic changes in pO2 or cyclic hypoxia. A significant difference from O2 diffusion-limited or chronic hypoxia is that the fluctuating hypoxic environment may directly influence the tumor vasculature itself, and the reoxygenation phases complicate the usual hypoxia-induced phenotypic pattern. Hypoxia induces some genes responsible for increased invasion, aggressiveness, and metastasis of tumors, including genes related to ECM interactions, migration, and proliferation [217]. Within a given tumor, there was an inverse correlation between regions of proliferation (Ki-67) and regions of hypoxia. The relationships between hypoxia and other biologic endpoints are complicated, but, within a given tumor's spatial relationships, they are in accord with known physiologic principles. Necrosis, proliferation, and blood vessel distribution cannot predict the level or presence of hypoxia in an individual tumor [218]. The exposure of endothelial cells to cycles of hypoxia/reoxygenation led to the accumulation of HIF-1alpha during the hypoxic periods and the phosphorylation of protein kinase B (Akt), extracellular regulated kinase (ERK) and endothelial nitric oxide synthase (eNOS) during the reoxygenation phases. Cyclic hypoxia, as reported in many tumor types, as a unique biological challenge for endothelial cells that promotes their survival in an HIF-1alpha-dependent manner through phenotypic alterations occurring during the reoxygenation periods [219].
Hypoxic tumors appear to be poorly differentiated. Increasing evidence suggests that hypoxia has the potential to inhibit tumor cell differentiation, thus playing a direct role in the maintenance of CSCs, also blocks differentiation of mesenchymal stem/progenitor cells, a potential source of tumor-associated stromal cells. It is therefore likely that hypoxia may have a profound impact on the evolution of the tumor stromal microenvironment, facilitating tumor progression. Hypoxia may help create a microenvironment enriched in poorly differentiated tumor cells and undifferentiated stromal cells. Such an undifferentiated hypoxic microenvironment may provide essential cellular interactions and environmental signals for the preferential maintenance of CSCs [220]. Hypoxia dramatically influences cellular phenotypes by altering the expression of specific genes, makes the tumors more aggressive (Figures 1), and is a significant contributor to intra-and inter-tumor cell diversity as revealed by the apparent but non-uniform expression of hypoxia-driven genes in solid tumors [221]. Hypoxic tumor cells lose their differentiated gene expression patterns and develop stem cell-like, immature or dedifferentiated phenotypes. Hypoxia-induced dedifferentiation will not only contribute to tumor heterogeneity but could also be one mechanism behind increased aggressiveness of hypoxic tumors. A state of hypoxia is a universal characteristic of the microenvironment of solid tumors. Growing evidence indicates that hypoxia plays a critical and fundamental role in metastasis formation [222]. Hypoxia drives tumor progression by influencing both the tumor cells as well as the tumor microenvironment (Figure 1). It increases genomic instability and heterogeneity in tumor cells and selects resistant tumor variants. It also alters the expression of several genes, among them hypoxia-inducible factor 1, a transcription factor that regulates a broad variety of the primary genes controlling cell survival, migration, and angiogenesis to name just a few [223,224]. Hypoxia and hypoxia-inducible factor 1 mediate progression-promoting or progression-restraining effects on tumor cells and the microenvironment. These activities involve, on the one hand, regulation of genes that drive tumor progression [223,224], and, on the other hand, the induction of genes that may cause the opposite effect. (e.g., cell death). By surviving and propagating under hypoxia, resistant tumor cells further aggravate the state of tumor hypoxia. This tumor expansion, in turn, increases genomic instability and further stabilizes and activates hypoxia-inducible factor 1. This vicious cycle drives alterations of gene expression patterns in tumor cells and thereby enhances tumor progression.
Hypoxia promotes each step of the metastatic cascade, including the promotion of angiogenesis and lymphangiogenesis, the induction of changes in vascular integrity and permeability (that facilitate tumor cell intravasation and extravasation), as well as cell proliferation [1]. Most of these mechanisms are regulated by HIF-induced VEGF action, which becomes connected with the coexpression through pVHL and might enable tumor progression and metastatic dissemination by autocrine receptor stimulation [225]. Hypoxia dramatically influences cellular phenotypes by altering the expression of specific genes, makes the tumors more aggressive, and is a significant contributor to intra-and inter-tumor cell diversity as revealed by the apparent but non-uniform expression of hypoxia-driven genes in solid tumors [221]. Intratumoral hypoxia is an independent indicator of poor patient outcome, and increasing evidence supports a role for hypoxia in the development of metastatic disease [221]. Studies suggest that the acquisition of the metastatic phenotype is not merely the result of dysregulated signal transduction pathways, but instead is achieved through a stepwise selection process driven by hypoxia [226,227,228]. Through up-regulation of urokinase-type plasminogen activator receptor (uPAR) expression [229], hypoxia enhances proteolytic activity at the invasive front. It also alters the interactions between integrins and components of the ECM, thereby enabling cellular invasion through the basement membrane and the underlying stroma [225,230,231]. Cell motility is increased through hypoxia-induced hepatocyte growth factor (HGF)-MET receptor signaling, resulting in cell migration towards the blood or lymphatic microcirculation. Hypoxia-induced vascular endothelial growth factor (VEGF) activity also plays a critical role in the dynamic tumor-stromal interactions required for the subsequent stages of metastasis [232]. HIF1-induced VEGF promotes angiogenesis and lymphangiogenesis, providing the necessary routes for dissemination, induces changes in vascular integrity and permeability that increase both intravasation and extravasation, and is essential for cell proliferation in metastatic lesions.
The necessary steps that enable metastasis are reversible, and therefore cannot be explained solely by irreversible genetic alterations, indicating the existence of a dynamic component to human tumor progression, in particular, a regulatory role for the tumor microenvironment [167]. Hypoxic tumor cells lose their differentiated gene expression patterns and develop stem cell-like, immature or dedifferentiated phenotypes [91]. Hypoxia-induced dedifferentiation will not only contribute to tumor heterogeneity but could also be one mechanism behind increased aggressiveness of hypoxic tumors. The loss of epithelial characteristics (i.e., lack of intercellular adhesion molecules) results in the breakdown of epithelial cell homeostasis, correlates with the acquisition of a migratory and stem cell phenotype that leads to progression, and it is regulated by CXCR4–CXCL12 [233]. The epithelial to mesenchymal transition is considered to be a crucial event in malignancy [234]. Tumors are morphological and functionally heterogeneous and segregate tumor cell with different capabilities: Individual tumor shows distinct sub-areas of proliferation and cell-cycle arrest, differentiation, cell adhesion and dissemination, some of them determined by topography [1,8,9,10,44,52,53,64,69,213,235]. Supporting this concept, topographic analysis of HIF1 and stem cell markers in follicular thyroid neoplasms has shown a direct correlation. Hypoxia facilitates disruption of tissue integrity through repression of E-cadherin expression, with a concomitant gain of N-cadherin expression, which allows cells to escape anoikis [1]. Neoplastic transformation evolves over a period involving the phenotypic progression of tumor cells along with the interaction of the initiated cell with its microenvironment. Tumor cell dissemination is the prerequisite of metastases and is correlated with loss of epithelial differentiation and the acquisition of a migratory phenotype through a stepwise, irreversible accumulation of genetic alterations [166,169,234]. The molecular mechanisms led by hypoxia explain the general phenotype associated with progression (metastatic capability): Spindle-shaped cellularity, necrosis, and stem cell phenotype. CXCR4–CXCL12 and hypoxia play a central role in the metastatic process (stromal interaction and digestion, angiogenesis), maintaining stem cell features in the tumor cells. Metastasis is considered the final step in malignancy that results from selected aspects of the complicated tumor progression process. However, this capability can be acquired early during the natural history of neoplasms and has a sophisticated molecular regulation, for which is primarily the interaction tumor cell-stroma. The chemokine axis CXCR4–CXCL12 has demonstrated a central role in this mechanism [236,237], involving both endothelial cells interactions and expression of metalloproteinases [225,229,238]. The pivotal role of hypoxia in tumor progression-metastasis would also explain the general phenotype associated with progression (metastatic capability): Spindle-shaped cellularity, necrosis, and stem cell phenotype. CXCR4–CXCL12 and hypoxia play a central role in the metastatic process (stromal interaction and digestion, angiogenesis), maintaining stem cell features in the tumor cells. Among the chemokines involved in tumor metastasis, the CXCL12 (also known as SDF-1 or stromal-derived factor)/CXCR4 axis plays a critical role in stem cell migration. Activation of CXCR4 induces motility, chemotactic response, adhesion, secretion of MMPs and release of angiogenic factors, such as VEGF-A. Interestingly this premetastatic niche, like the usual niche, is characterized by the presence of specific ECM proteins, such as fibronectin, a ligand for α4β1 expressed on VEGFR-1+ cells. Thus, it appears that to survive at distant sites, disseminating tumor cells need to recreate a supportive microenvironment similar to the one formed in the primary tumor. One efficient way to do it is by directing BMDC or immune/inflammatory cells to these distant sites of future metastasis [239]. The chemoattractant proteins S100A8 and S100A9 were the first factors shown to instruct the formation of the pre-metastatic niche [172,240]. Additional studies are needed to elucidate further the biological mechanisms involved in the formation of this premetastatic niche, in particular regarding the identification of the molecular mechanism responsible for the development of clinically relevant metastases from the first seeds.
The phenotypic heterogeneity in neoplasms is mainly related to differential gene expression. Gene expression analysis is becoming a useful tool for a better definition of neoplasms at diagnostic, prognostic and predictive levels. The identification of predictive markers of these features will help both classifying neoplasms and stratifying patients for better management. However, the nature and biological meaning of these gene expression markers are not always evident: The origin of the tested RNA, mechanism of RNA transference, a utility for subclassification of neoplastic lesions.
The mutational landscape of cancer is complex and individual cancers may evolve through mutations in as many as 20 different cancer-associated genes. Gene expression analysis is becoming a useful tool for a better definition of neoplasms at diagnostic, prognostic and predictive levels [241,242,243,244,245]. Tumor hypoxia induces changes in the proteome and genome of neoplastic cells that enable the cells to overcome nutritive deprivation or to escape their hostile environment. The selection and clonal expansion of these favorably altered cells further aggravate tumor hypoxia and support a self-perpetuating circle of increasing hypoxia and malignant progression while concurrently promoting the development of more treatment-resistant disease [246]. The Met tyrosine kinase, a high-affinity receptor for hepatocyte growth factor (HGF), plays a crucial role in controlling invasive growth and is often overexpressed in cancer. Hypoxia activates MET protooncogene transcription, amplifies HGF (hepatocyte growth factor) signaling, and synergizes with HGF in inducing invasion; the pro-invasive effects of hypoxia are mimicked by Met overexpression (Figure 1). Hypoxic tumor areas overexpress Met, and the inhibition of Met expression prevents hypoxia-induced invasive growth. These data show that hypoxia promotes tumor invasion by sensitizing cells to HGF stimulation, providing a molecular basis to explain Met overexpression in cancer [247]. There is also a link between a specific group of microRNAs and hypoxia, a vital feature of the neoplastic microenvironment. A significant proportion of the hypoxia-regulated microRNAs is also overexpressed in human cancers, suggesting a role in tumorigenesis, affecting critical processes such as apoptosis, proliferation, and angiogenesis, like the induction in response to HIF activation [248].
The gene expression signature also reflects the metastatic potential in both cell lines and tumor samples [249,250,251,252,253]. Some general conclusions have been drawn in this respect. First, the expression profile of low metastatic subline is distinct from that of the high metastatic subline. Metastases of the low metastatic subline closely resembled the profile of the low metastatic primary tumor and did not "switch over" to the high metastatic gene expression profile. Thus, heterogeneity exists in this cell line/tumor, but may not be apparent as the signature reflects the majority of the tumor cells. The low metastatic subline is capable of spawning metastases, albeit at a lower rate, and its gene expression profile is therefore of significant interest. Second, the gene expression profile of the sublines as in vitro cultures is very distinct from the same sublines as primary tumors. While the heterotypic biology of primary tumors, the contribution of their constituent cells to the observed gene expression profiles should be minimal. Thus, the differences are thought to reflect changes in the tumor cells in response to the in vivo microenvironment. Third, comparable numbers of genes were "turned on" and "turned off" in the more highly metastatic tissues. We think about metastasis as the acquisition of traits, but the data remind us to pay equal attention to the loss of growth and differentiation-controlling genes. Many of the differentially expressed genes fall into the category of "usual suspects" for metastasis. These include the overexpression of osteopontin and ECM genes in the more metastatic tissues and the loss of thrombospondin 1 and members of the Nm23 family [243]. Eight metastasis-suppressor genes that reduce the metastatic propensity of a cancer cell line in vivo without affecting its tumorigenicity have been identified. These affect critical signal transduction pathways, including mitogen-activated protein kinases, RHO, RAC and G-protein-coupled and tyrosine-kinase receptors [251]. Other genes are unexpected and, if validated functionally, could shed new light on the mechanisms of metastasis.
The biologic heterogeneity of intraepithelial and invasive malignancies is well known at the morphologic, kinetic and genetic levels, an issue that has not been addressed in this paratumoral gene expression analysis and should warrant future studies. The gene expression markers should be evaluated in the appropriate biological context. Gene expression profiles are determined by a gene regulatory network comprising the regulatory core of genes represented most prominently by transcription factors and miRNAs, which influence the expression of other genes, and a periphery of effector genes that are regulated but not regulating [254,255]. Most studies do not differentiate between these two essential groups, which can also be useful in selecting surrogate markers for a given condition [1,254]. There is a general concept to keep in mind for gene expression analyses: The amount of information is overwhelming, and the number of variables included in the studies significantly outnumbers the cases. In this scenario, the significant variables can be the result of a statistical selection rather than the expression of a biologically significant process for a particular neoplasm. Significant variables frequently include genes of the general metabolic activation associated with the neoplastic transformation [1,2], rather than tissue-or differentiation-specific gene variables.
The application of the so-called hallmarks of cancer in oncological pathology leads to consideration of the molecular test requirements (Molecular Test Score System) for reliable implementation; these requirements should cover biological effects, molecular pathway, biological validation, and technical validation [1]. Sensible application of molecular markers in tumor pathology always needs a solid morphological support [1,62].
Additionally, any new test should be validated against the accepted standard (specificity/sensitivity), demonstrate improvements in patient's management, and should provide a biologic meaning for its application. These requirements could be considered as implementation phases: The initial design typically meets the first requirement, the second requirement would be expected in any successful implementation, while the third is harder to prove. Any new definition should be biologically meaningful and would incorporate core elements in tumor biology (in particular genetic and kinetic correlates). Apart from the lack of general interest, the scarcity of definitive publications on clonal heterogeneity can be attributed to the substantial technical challenges faced by researchers who want to study genetic heterogeneity in human cancers. Several approaches have been used to examine genetic heterogeneity, which needs to address the issue of small sampling and technical caveats arising from the frequent necessity to analyze limited amounts of starting material. It is useful to divide the approaches to study clonal heterogeneity into focused and genome-wide methods.
Tumor progression assessment would ideally analyze at least two samples to compare the biologic markers relevant for progression in both tumor cell clones and the microenvironment. The tumor clone markers include those involved in the tumorigenic expansion (proliferation), and invasion, the two leading forces driving progression. The tumor microenvironment analysis focuses the attention on the qualitative that potentiate clonal expansion and invasion of tumor cells. In essence, the tumor progression analysis has to concentrate on clonal heterogeneity and overcome its problems. One aspect of this matter is the availability of representative biopsies. Core biopsies sample only small regions of tumors, and, therefore, are not likely to be informative about the clonal composition of each tumor as a whole [9,10,11,52,53,61,213]. The sample size is another critical issue [45,46,58]. On sample size, there is a need to balance signal-to-noise ratios. Many studies achieve this balance by focusing on relatively small regions of tumors, ideally representing morphologically distinct units that are isolated by microdissection. The validity of this approach is supported by the observation of the clustering of populations that differ in gene expression [256], as well as genetic composition [214]. However, sampling small regions of tumors can still miss relatively small patches of genetically distinct cells, unless large numbers of samples are taken from the individual tumors [45,46,58]. Also, if genetically distinct clones are intermixed within small anatomically distinct units, such heterogeneity will be missed due to pooling of cells.
The deep sequencing of whole genomes allows for the detection of mutant alleles present in a small fraction of tumor cells [183] and can be performed with starting material of just single cells [190]. Focusing on single cells, however, brings about the problem of dealing with cell-to-cell variability, where some of the distinguishable variants have no chances of clonal expansion and thus constitute evolutionary "dead ends". This problem of biological noise can potentially be dealt with by analyzing more substantial numbers of individual cells, but scaling up the analysis will inevitably drive up the cost and labor required, making these studies impractical. Moreover, single-cell analysis suffers from increased vulnerability to technical caveats related to the necessity to amplify single genomes before the analysis. Another approach to sampling is based on the analysis of phenotypically distinct subpopulations after singling them out by FACS, which can reveal the existence of unexpected clonal divergence between the two populations in some tumor samples [257]. Obviously, this approach can be valid only for the cases when phenotypic differences can be related to differences in genetic composition, and genetic heterogeneity within the populations will be missed.
The distinct disadvantage of these methods is that limiting the analysis to a few loci will miss other potentially significant differences and will likely underestimate the extent and patterns of clonal heterogeneity. Current knowledge of cancer genetics permits focusing on genetic lesions specific for types and subtypes of particular cancer. Specific genetic perturbations have been implicated in initiation and progression. However, to a great extent, underlying mechanisms often remain elusive. These genetic perturbations are most likely reflected by the altered expression of sets of genes or pathways, rather than individual genes, thus creating a need for models of deregulation of pathways to help provide an understanding of the mechanisms of tumorigenesis. The use of gene sets in modeling tumor progression rather than single genes is a useful approach taking advantage of the available information for specific neoplasms. Cancer progression has taken a step closer to gaining insight into biologically related gene sets implicated throughout disease by the identification of relevant cellular functions based on shared characteristics of the individual genes found to be differentially expressed. Using defined gene sets relevant at different stages of the disease and the interaction network of these relevant gene sets, appropriate pathways and genes most relevant to progression can be developed, along with their corresponding interactions [1,258]. This approach provides the foundations for the biological analysis of the capabilities involved in progression as promoting (proliferation and invasion) or suppressive (cytostasis-differentiation and viability) factors.
These approaches allow for the analysis of specific genes that have been implicated as drivers of tumor progression. Clonal heterogeneity can be assessed by sequencing specific genes known to be frequently mutated in particular cancer [214,259]. This analysis usually involves microdissection and PCR amplification of the samples. The significant advantage of sequencing is that it allows for focusing on specific "driver" genes; thus, any heterogeneity will most likely be of functional importance. Intensive studies in the field of cancer genetics, and recent sequencing of cancer genomes and allowed the identification of cancer-type-specific subsets of genes that "drive" cancer progression [257,260,261]. Therefore, it should be feasible to analyze samples for sets of genes causally implicated in a given cancer. Also, instead of the detection of specific nucleotide changes, bisulfite sequencing of specific alleles can be used to detect heritable methylation changes either in promoters of "driver" genes or in a larger set of "passenger" sites, which can be used as markers to trace clonal expansion patterns [262]. Combining PCR and deep sequencing appears to be a particularly promising way to analyze the evolutionary relationship among distinct clones, as it allows for the detection of rare subclones without a priori knowledge of the products of rearrangements [113,263]. One potential drawback is that the subclonal changes might have no effect on fitness, i.e., they are evolutionarily neutral. However, the approach is still useful given that it allows for establishing evolutionary relationships between primary disease and relapses [264,265].
Most commonly, allelic imbalance analysis is performed on microdissected tumor samples to ensure lack of contamination by healthy cells and to provide adequate sample sizes. The DNA is then isolated from the captured cells, and PCR is performed using primers against microsatellite markers known to be frequently altered in the particular type or subtype of cancer under study. Comparison to Controls, germline samples from the same patients, allows for the detection of alleles that have been lost somatically. Although obtaining data is relatively straightforward, straightforward interpretation is obscured by various technical caveats, such as the frequently poor correlation between chromosomal deletion/amplification and under/over representation of significant microsatellite markers [266]. Therefore, data obtained by analysis of allelic imbalances often requires confirmation by independent techniques.
Studies of clonal heterogeneity may rely on the detection of amplification of specific chromosomal regions by fluorescent in situ hybridization (FISH) or immuno-FISH (FISH coupled with immunofluorescence) analysis [257,267]. In the FISH analysis, fluorescently labeled BAC (bacterial artificial chromosome) probes against regions amplified in the particular cancer type, as well as control probes for centromeric regions, are hybridized to cancer cells or tissue sections. The combination with immunofluorescence provides the advantage of focusing on specific subtypes of tumor cells. The FISH analysis is less vulnerable to artifacts than are other approaches, such as analysis of allelic imbalances; thus, it can be the approach of choice for the validation of differences initially detected by other means. Disadvantages include its labor-intensiveness, as a large number of cells need to be analyzed to generate statistically meaningful data. Also, although potentially suitable for the analysis of chromosomal deletions, detection of amplified regions is more practical.
Lymphoid malignancies sometimes present another unique opportunity for focused clonality analysis. Early steps of development of B and T cells involve step-wise rearrangements of immunoglobulin (Ig) and T-cell receptor (TCR) genes, respectively. Germline loci for Ig and TCR genes contain multiple alternative gene segments, encoding for gene domains, that are randomly chosen for rearrangements to generate thousands to millions (depending on the gene) potential mature rearranged products distinguishable by PCR [268,269].
A distinct advantage of these approaches is that their success is not contingent on a priori knowledge; therefore, potentially significant differences, missed by focused approaches can be detected. The significant disadvantages of these methods are that the functional impact of detected differences is often unclear and that the clonal differences might be evolutionarily neutral. Also, many genome-wide approaches suffer from limited resolution.
Karyotypic analysis based on chromosomal banding was frequently used in early reports on clonal heterogeneity [270,271]. In this technique, tumor cells are subjected to short-term culture followed by fixation and staining, which reveals a pattern of chromosomal banding in metaphase chromosomes. This method allows for the detection of the primary chromosomal abnormalities, but it has several pitfalls. Karyotypic analysis requires culturing the cells, which may lead to preferential outgrowth of selected tumor cell subpopulations, changing the representation of the original tumor. It is also labor-intensive and has low resolution.
More recent publications usually rely on comparative genomic hybridization (CGH) to detect chromosomal aberrations. DNA from tumor and normal cells are differentially labeled with fluorescent probes and, in the classical form of the assay, hybridized to normal metaphase chromosomes. The fluorescent signal is then analyzed using specialized software, revealing regions of chromosomal losses and gains. CGH is one of the most widely used techniques in studies of intra-tumor clonal heterogeneity. It provides unbiased coverage of the whole genome. However, hybridization to metaphase chromosomes gives limited resolution, as small regions of deletion and amplification can be missed. Instead of metaphase chromosomes, differentially labeled DNA can be hybridized to genomic arrays, allowing for the detection of smaller-scale allelic imbalances up to the single-nucleotide level using single nuclear polymorphism (SNP) arrays [257]. Notably, analysis of chromosomal imbalances can be performed on the level of specific chromosomes (using chromosomal arrays) rather than on a whole-genome scale. Finally, clonal heterogeneity can potentially be detected by the analysis of DNA content, where tumor cells are stained with DNA-labeling dye and subjected to flow cytometric analysis (FACS). This approach reveals ploidy status and the distribution of cells in different phases of the cell cycle. In cases of substantial differences, it allows for the detection of populations with distinct aneuploidy based on their shift from regular ploidy peaks [49,272,273,274]. The advantages of this method are that sampling issues are not much of a problem and that it is capable of resolving even highly intermixed subpopulations. However, its resolution is limited to the detection of clones that differ substantially in DNA content. Therefore, the method provides limited molecular insights unless distinct subpopulations are sorted and subsequently analyzed with more sensitive methods.
Many methods of tumor heterogeneity analysis rely on microdissection of small, anatomically distinct sections of tumors from frozen or archived tissues. Microdissection allows for focusing the analysis on small, morphologically homogeneous populations and avoids contamination with non-malignant cells. However, the technique leads to increased DNA damage and thus reduced quality. The issue of DNA quality is especially pronounced in the analysis of archived formalin-fixed paraffin embedded tissues. Clonality remains as one of the most important hallmarks of neoplasms, but its analysis requires particular test designs. The core of comprehensive clonality assays would include a complementary approach comprising the evaluation of structural abnormalities of crucial oncogenes and tumor suppressor genes (mutations, allelic imbalance) that control essential acquired capabilities in neoplasms. Pathway independent tests based on X-chromosome inactivation and the evaluation of epigenetic alterations such as methylation levels have a more restrictive application in female and specific tumor types with high imprinting levels, respectively. This approach would resolve the patch size problem that is frequently associated with clonal expansions, which can be highlighted in single marker analyses.
Due to the low amount of starting material, many tumor heterogeneity analysis methods, especially those involving whole-genome analysis, rely on prior amplification of whole genomes. Several efficient methods for whole-genome amplification, relying either on PCR-based or isothermal DNA amplification, have been developed over the last decades. However, whole-genome amplification produces only a representation, not a replica, of the genome, so, inevitably, representation bias is introduced. This bias might be more pronounced in PCR-based techniques due to different efficiencies of amplification of fragments of various sizes and nucleotide composition. However, even isothermal, linear amplification techniques have been shown to suffer from substantial representation bias [275]. The issue of representation bias is most pronounced when the starting material is small (<10 cells) [46,58], culminating at the single-cell level. To some extent, bias issues can be reduced by using identically manipulated control samples. However, acquiring robust data requires confirmation by independent methods, such as FISH analyses.
It is possible that measurements of clonal heterogeneity can be used irrespective of their utility in routine diagnostics. Substantial clonal heterogeneity between primary and metastatic tumors can pose a barrier to targeted therapy. Since biopsies of metastases are frequently unavailable, it would be worth investigating if the clonal heterogeneity of primary tumors is indicative of clonal divergence in metastatic outgrowths. The index of clonal diversity is a strong predictor of malignant progression in Barrett's esophagus [49]; thus, it can be used for guiding treatment decisions. Although experimental evidence is still lacking, it is likely that Barrett's esophagus is not a biological oddity and that clonality measurements will turn out to be of predictive value for malignant progression in other cancers as well. However, sampling multiple regions from the same tumor are impractical for most cancer types, and it is not yet clear if obtaining accurate clonality measurements can be achieved using current routine diagnostic methods. Also, though multiple biopsies from a single tumor might be impractical, the degree of cellular and molecular heterogeneity within a relatively small sample might also turn out to be of predictive value.
Biological interactions between tumor clones could have a dramatic impact on tumor evolution, both in a natural progression and an evolution in response to selection forces created by therapeutic interventions. This suggestion is supported by experimental studies in mice that have demonstrated that the growth of tumors resulting from transplanting mixtures of clones cannot be predicted from the behavior of tumors developed from individual clones [276]. Similarly, experimental tumors composed of multiple clones display different sensitivity to cytotoxic drugs compared to monoclonal tumors, as clonal interactions can either potentiate or inhibit therapeutic efficacy. Therefore, clonal heterogeneity adds a level of complexity to our understanding of the biology of tumor development and poses challenges for the development of successful therapies. As the field of cancer treatment is moving toward the development of targeted therapeutic approaches, the question of whether the expression of a therapeutic target in a biopsy is representative of the whole corresponding tumor becomes of primary importance [277]. For example, clonal heterogeneity involving different mutational statuses was documented for KRAS [272], tyrosine kinase growth factor receptors such as EGFR [278,279], and TP53 [49,280] genes, for which targeted therapy is currently being developed. Heterogeneous expression of the therapeutic target does not necessarily mean that the tumor will not be responsive to the treatment, especially if the targets are responsible for the "common good". For example, targeting an angiogenic clone can also eliminate non-angiogenic "free rider" clones, which are dependent on the secretion of angiogenic factors. Still, clonal heterogeneity needs to be accounted for in the development of targeted therapy approaches, considering that high genetic heterogeneity of tumors means a high probability of pre-existent clones that are resistant to therapeutic intervention and can be selected for by treatment, resulting in therapy failure.
On the other hand, clonal heterogeneity can potentially be exploited for therapeutic benefit. Maley et al. proposed two such strategies of anti-cancer therapy. First, boosting the selective advantage of non-malignant clones can be used to drive malignant clones to extinction. Second, the selective advantage of clones sensitive to a particular therapy can be boosted, allowing them to achieve clonal dominance, and then treatment can be applied. In silico modeling confirmed that these strategies could be much more efficient compared to standard cytotoxic therapies [281]. Unfortunately, these strategies are not yet feasible, as we lack the knowledge of how to single out or boost non-malignant or chemosensitive clones. Gatenby and co-authors have recently proposed another strategy, termed adaptive therapy, which both accounts for the heterogeneity of tumor cells and takes advantage of it [215]. The conventional approach of adjuvant therapy is to hit tumors with the highest tolerated dose of cytotoxic drugs in a hope to kill the most significant possible numbers of tumor cells, thereby decreasing the probability of the emergence of resistant mutants. However, this strategy would be expected to fail if resistant clones already exist before the treatment starts. Indeed, despite initial therapeutic responsiveness, treatment almost inevitably ends with the outgrowth of resistant tumors. Instead of completely eradicating tumors (which is not practically achievable in most cases), adaptive therapy aims to keep tumor size at bay by adjusting drug dose and timing of drug administration based on tumor response. This strategy prevents resistant variants from reaching fixation by maintaining the dominance of therapy-sensitive variants. Importantly, the feasibility of adaptive therapy is not contingent on knowledge of the exact mechanisms of the resistance, though the applicability of this idea has yet to be confirmed.
Elimination of MDSC is a priority for cancer patients who are candidates for active immunotherapy. Likewise, limiting the accumulation and retention of MDSC during chronic inflammation may reduce the risk of developing cancers. The proinflammatory mediators that induce MDSC are particularly attractive targets for limiting this suppressor cell population, although several unknowns make it difficult to decide which mediators to target. For example, it is not known whether the various pro-inflammatory factors induce MDSC through independent or overlapping pathways. If the pathways are independent, then it will be necessary to block particular pathways. In contrast, if the pathways merge, then a single drug aimed at a typical molecule may be sufficient. Future studies may identify additional proinflammatory mediators or factors that independently or coordinately regulate the accumulation of MDSC. Regardless of the complexity of MDSC induction, reduction of this inhibitory population is essential, and a comprehensive understanding of the proinflammatory mediators that regulate MDSC will provide valuable information for future drug design.
Co-existence of distinct clones within a tumor can have profound clinical implications for disease progression, diagnosis, and therapeutic responses. Both tumor progression and intratumoral heterogeneity are essential to understanding the response to therapy, and they will be better evaluated by targeting several pathways [278,282]. This practical aspect is crucial to a more efficient personalized therapy, but there are biological aspects that would be worth considering. The neoplastic transformation evolves over a period involving the phenotypic progression of tumor cells along with the interaction of the initiated cell with its microenvironment (Figure 3). The elucidation of the steps of cancer progression relates to the precarious balance of acquired capabilities of cell proliferation, invasion, viability, and cytostasis/differentiation. This balance is of utmost importance in the differential diagnosis of neoplasms and the establishment of more efficacious therapeutic regimens. This functional characterization of the particular stage of the tumor will undoubtedly allow for the better diagnosis, staging, prognostication, and treatment of cancers. The landscaper effect of the microenvironment and its transition from tissue to tumor variants determine a qualitative change be enhancing instability and enabling progression.
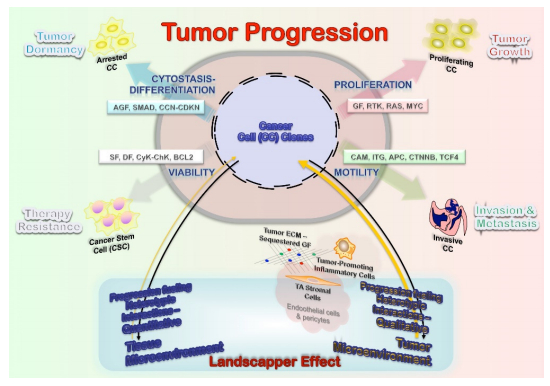 Figure 3. Essential interactions are likely to exist between distinct tumor clones, these biological interactions are not mutually exclusive, and the net outcome will depend on the net sum of the different interactions of gene clusters. Inhibiting effects would result from progression interfering interactions (competition, amensalism, antagonism) while stimulating effects are the consequence of progress boosting interactions (commensalism, mutualism). The overall tumor growth and progression can be both augmented and retarded by these forces.
Figure 3. Essential interactions are likely to exist between distinct tumor clones, these biological interactions are not mutually exclusive, and the net outcome will depend on the net sum of the different interactions of gene clusters. Inhibiting effects would result from progression interfering interactions (competition, amensalism, antagonism) while stimulating effects are the consequence of progress boosting interactions (commensalism, mutualism). The overall tumor growth and progression can be both augmented and retarded by these forces.Significant amounts of evidence support the close clonal relationship of primary tumors and metastatic outgrowths. However, there is also substantial evidence that suggests that metastatic spread can be an early event during tumor progression; thus, primary and metastatic tumors might become quite distinct genetically as they evolve. Some of the observed discrepancies might stem from technical issues, but it is plausible that the development of spontaneous human tumors is not confined to a single scenario. It is also possible that while many tumor cells are capable of early dissemination, only tumor cells that have acquired a significant number of oncogenic mutations are capable of initiating secondary tumors at metastatic sites. A stochastic multi-gene approach that covers the acquired capabilities and builds the relevant pathways promoting progression (proliferation and invasion) of viable tumor cells would be the most sensible biological evaluation.
Comprehensive characterization of tumor clonality is contingent on the development of approaches that can combine a focus on "driver" lesions with large-scale analysis. It is needed a more systemic approach to characterize the extent of clonal heterogeneity of different types and subtypes of cancers through various stages of tumor development, including disseminated tumor cells and metastases, and response to treatment. Advances in our understanding of the genetic composition of cancers make these analyses potentially feasible; however, methodology allowing for robust and cost-efficient analyses of small tumor samples is still lacking.
Accounting for the biological interactions among distinct clonal populations within tumors might pose the biggest challenge. The co-existence of multiple clones within a tumor leads to a complex network of interactions both among the clones and between the clones and the tumor microenvironment. Thus, tumors are likely to behave as complex systems and should be treated as such. While deciphering the full extent of the interactions occurring within tumors may be an impossible task, we might be able to identify critical links and use this knowledge to control or eliminate tumors. The knowledge from the fields of evolutionary biology and ecology provides critical insights for improving the clinical management of cancer patients.
All authors declare no conflicts of interest in this paper.
| [1] | Diaz-Cano SJ (2008) General morphological and biological features of neoplasms: Integration of molecular findings. Histopathology 53: 1–19. |
| [2] |
Hanahan D, Weinberg RA (2011) Hallmarks of cancer: The next generation. Cell 144: 646–674. doi: 10.1016/j.cell.2011.02.013

|
| [3] |
Hanahan D, Weinberg RA (2000) The hallmarks of cancer. Cell 100: 57–70. doi: 10.1016/S0092-8674(00)81683-9
|
| [4] |
Kinzler KW, Vogelstein B (1996) Lessons from hereditary colorectal cancer. Cell 87: 159–170. doi: 10.1016/S0092-8674(00)81333-1
|
| [5] |
Fidler IJ, Hart IR (1982) Biological diversity in metastatic neoplasms: Origins and implications. Science 217: 998–1003. doi: 10.1126/science.7112116
|
| [6] | Heppner GH, Miller FR (1998) The cellular basis of tumor progression. Int Rev Cytol 177: 1–56. |
| [7] |
Talmadge JE, Wolman SR, Fidler IJ (1982) Evidence for the clonal origin of spontaneous metastases. Science 217: 361–363. doi: 10.1126/science.6953592
|
| [8] |
Blanes A, Diaz-Cano SJ (2006) Complementary analysis of microsatellite tumor profile and mismatch repair defects in colorectal carcinomas. World J Gastroenterol 12: 5932–5940. doi: 10.3748/wjg.v12.i37.5932
|
| [9] |
Blanes A, Diaz-Cano SJ (2006) DNA and kinetic heterogeneity during the clonal evolution of adrenocortical proliferative lesions. Hum Pathol 37: 1295–1303. doi: 10.1016/j.humpath.2006.04.025
|
| [10] |
Blanes A, Sanchez-Carrillo JJ, Diaz-Cano SJ (2006) Topographic molecular profile of pheochromocytomas: Role of somatic down-regulation of mismatch repair. J Clin Endocrinol Metab 91: 1150–1158. doi: 10.1210/jc.2005-1645
|
| [11] | Pozo L, Sanchez-Carrillo JJ, Martinez A, et al. (2007) Differential kinetic features by tumour topography in cutaneous small-cell neuroendocrine (Merkel cell) carcinomas. J Eur Acad Dermatol Venereol 21: 1220–1228. |
| [12] |
Kinzler KW, Vogelstein B (1998) Landscaping the cancer terrain. Science 280: 1036–1037. doi: 10.1126/science.280.5366.1036
|
| [13] |
Parmigiani G, Boca S, Lin J, et al. (2009) Design and analysis issues in genome-wide somatic mutation studies of cancer. Genomics 93: 17–21. doi: 10.1016/j.ygeno.2008.07.005
|
| [14] |
Polyak K, Haviv I, Campbell IG (2009) Co-evolution of tumor cells and their microenvironment. Trends Genet 25: 30–38. doi: 10.1016/j.tig.2008.10.012
|
| [15] |
Nowell PC (1976) The clonal evolution of tumor cell populations. Science 194: 23–28. doi: 10.1126/science.959840
|
| [16] |
McGranahan N, Swanton C (2017) Clonal Heterogeneity and Tumor Evolution: Past, Present, and the Future. Cell 168: 613–628. doi: 10.1016/j.cell.2017.01.018
|
| [17] |
Shah NP, Nicoll JM, Nagar B, et al. (2002) Multiple BCR-ABL kinase domain mutations confer polyclonal resistance to the tyrosine kinase inhibitor imatinib (STI571) in chronic phase and blast crisis chronic myeloid leukemia. Cancer Cell 2: 117–125. doi: 10.1016/S1535-6108(02)00096-X
|
| [18] |
Corless CL, Heinrich MC (2008) Molecular pathobiology of gastrointestinal stromal sarcomas. Annu Rev Pathol 3: 557–586. doi: 10.1146/annurev.pathmechdis.3.121806.151538
|
| [19] |
Edwards SL, Brough R, Lord CJ, et al. (2008) Resistance to therapy caused by intragenic deletion in BRCA2. Nature 451: 1111–1115. doi: 10.1038/nature06548
|
| [20] |
Blagosklonny MV (2002) Oncogenic resistance to growth-limiting conditions. Nat Rev Cancer 2: 221–225. doi: 10.1038/nrc743
|
| [21] |
Laconi E, Doratiotto S, Vineis P (2008) The microenvironments of multistage carcinogenesis. Semin Cancer Biol 18: 322–329. doi: 10.1016/j.semcancer.2008.03.019
|
| [22] |
Laconi E, Sonnenschein C (2008) Cancer development at tissue level. Semin Cancer Biol 18: 303–304. doi: 10.1016/j.semcancer.2008.03.002
|
| [23] | Bissell MJ, Labarge MA (2005) Context, tissue plasticity, and cancer: Are tumor stem cells also regulated by the microenvironment? Cancer Cell 7: 17–23. |
| [24] |
Polyak K (2011) Heterogeneity in breast cancer. J Clin Invest 121: 3786–3788. doi: 10.1172/JCI60534
|
| [25] |
Merlo LM, Pepper JW, Reid BJ, et al. (2006) Cancer as an evolutionary and ecological process. Nat Rev Cancer 6: 924–935. doi: 10.1038/nrc2013
|
| [26] |
Axelrod R, Axelrod DE, Pienta KJ (2006) Evolution of cooperation among tumor cells. Proc Natl Acad Sci U S A 103: 13474–13479. doi: 10.1073/pnas.0606053103
|
| [27] |
Mallepell S, Krust A, Chambon P, et al. (2006) Paracrine signaling through the epithelial estrogen receptor alpha is required for proliferation and morphogenesis in the mammary gland. Proc Natl Acad Sci U S A 103: 2196–2201. doi: 10.1073/pnas.0510974103
|
| [28] |
Godar S, Ince TA, Bell GW, et al. (2008) Growth-inhibitory and tumor- suppressive functions of p53 depend on its repression of CD44 expression. Cell 134: 62–73. doi: 10.1016/j.cell.2008.06.006
|
| [29] |
McAllister SS, Gifford AM, Greiner AL, et al. (2008) Systemic endocrine instigation of indolent tumor growth requires osteopontin. Cell 133: 994–1005. doi: 10.1016/j.cell.2008.04.045
|
| [30] |
Nagy JD (2004) Competition and natural selection in a mathematical model of cancer. Bull Math Biol 66: 663–687. doi: 10.1016/j.bulm.2003.10.001
|
| [31] |
Yamamoto K, Hanada R, Kikuchi A, et al. (1998) Spontaneous regression of localized neuroblastoma detected by mass screening. J Clin Oncol 16: 1265–1269. doi: 10.1200/JCO.1998.16.4.1265
|
| [32] |
Axelrod R, Hamilton WD (1981) The evolution of cooperation. Science 211: 1390–1396. doi: 10.1126/science.7466396
|
| [33] |
Lyons JG, Lobo E, Martorana AM, et al. (2008) Clonal diversity in carcinomas: Its implications for tumour progression and the contribution made to it by epithelial-mesenchymal transitions. Clin Exp Metastasis 25: 665–677. doi: 10.1007/s10585-007-9134-2
|
| [34] | Marusyk A, DeGregori J (2008) Declining cellular fitness with age promotes cancer initiation by selecting for adaptive oncogenic mutations. Biochim Biophys Acta 1785: 1–11. |
| [35] |
Lowe SW, Cepero E, Evan G (2004) Intrinsic tumour suppression. Nature 432: 307–315. doi: 10.1038/nature03098
|
| [36] |
Zhang J, Grindley JC, Yin T, et al. (2006) PTEN maintains haematopoietic stem cells and acts in lineage choice and leukaemia prevention. Nature 441: 518–522. doi: 10.1038/nature04747
|
| [37] |
Michor F, Iwasa Y, Nowak MA (2004) Dynamics of cancer progression. Nat Rev Cancer 4: 197–205. doi: 10.1038/nrc1295
|
| [38] |
Michor F, Polyak K (2010) The origins and implications of intratumor heterogeneity. Cancer Prev Res 3: 1361–1364. doi: 10.1158/1940-6207.CAPR-10-0234
|
| [39] |
Marusyk A, Casas-Selves M, Henry CJ, et al. (2009) Irradiation alters selection for oncogenic mutations in hematopoietic progenitors. Cancer Res 69: 7262–7269. doi: 10.1158/0008-5472.CAN-09-0604
|
| [40] |
Laconi S, Pani P, Pillai S, et al. (2001) A growth-constrained environment drives tumor progression invivo. Proc Natl Acad Sci U S A 98: 7806–7811. doi: 10.1073/pnas.131210498
|
| [41] |
Devireddy LR, Gazin C, Zhu X, et al. (2005) A cell-surface receptor for lipocalin 24p3 selectively mediates apoptosis and iron uptake. Cell 123: 1293–1305. doi: 10.1016/j.cell.2005.10.027
|
| [42] | Guba M, Cernaianu G, Koehl G, et al. (2001) A primary tumor promotes dormancy of solitary tumor cells before inhibiting angiogenesis. Cancer Res 61: 5575–5579. |
| [43] |
Iwasa Y, Michor F (2011) Evolutionary dynamics of intratumor heterogeneity. PLoS One 6: e17866. doi: 10.1371/journal.pone.0017866
|
| [44] |
Diaz-Cano SJ (1998) Clonality studies in the analysis of adrenal medullary proliferations: Application principles and limitations. Endocr Pathol 9: 301–316. doi: 10.1007/BF02739690
|
| [45] |
Diaz-Cano SJ (2000) Designing a molecular analysis of clonality in tumours. J Pathol 191: 343–344. doi: 10.1002/1096-9896(200008)191:4<343::AID-PATH617>3.0.CO;2-Y
|
| [46] |
Diaz-Cano SJ, Blanes A, Wolfe HJ (2001) PCR techniques for clonality assays. Diagn Mol Pathol 10: 24–33. doi: 10.1097/00019606-200103000-00005
|
| [47] | Pozo-Garcia L, Diaz-Cano SJ (2003) Clonal origin and expansions in neoplasms: Biologic and technical aspects must be considered together. Am J Pathol 162: 354–355. |
| [48] |
Leedham SJ, Wright NA (2008) Human tumour clonality assessment-flawed but necessary. J Pathol 215: 351–354. doi: 10.1002/path.2379
|
| [49] |
Maley CC, Galipeau PC, Finley JC, et al. (2006) Genetic clonal diversity predicts progression to esophageal adenocarcinoma. Nat Genet 38: 468–473. doi: 10.1038/ng1768
|
| [50] |
Prasetyanti PR, Medema JP (2017) Intra-tumor heterogeneity from a cancer stem cell perspective. Mol Cancer 16: 41. doi: 10.1186/s12943-017-0600-4
|
| [51] |
Arif S, Blanes A, Diaz-Cano SJ (2002) Hashimoto's thyroiditis shares features with early papillary thyroid carcinoma. Histopathology 41: 357–362. doi: 10.1046/j.1365-2559.2002.01467.x
|
| [52] |
Blanes A, Rubio J, Martinez A, et al. (2002) Kinetic profiles by topographic compartments in muscle-invasive transitional cell carcinomas of the bladder: Role of TP53 and NF1 genes. Am J Clin Pathol 118: 93–100. doi: 10.1309/8QR9-2FRE-JPHF-VRC6
|
| [53] |
Diaz-Cano SJ, Blanes A, Rubio J, et al. (2000) Molecular evolution and intratumor heterogeneity by topographic compartments in muscle-invasive transitional cell carcinoma of the urinary bladder. Lab Invest 80: 279–289. doi: 10.1038/labinvest.3780033
|
| [54] |
Diaz-Cano SJ, Miguel MD, Blanes A, et al. (2000) Clonality as expression of distinctive cell kinetics patterns in nodular hyperplasias and adenomas of the adrenal cortex. Am J Pathol 156: 311–319. doi: 10.1016/S0002-9440(10)64732-3
|
| [55] |
Diaz-Cano SJ, Miguel MD, Blanes A, et al. (2000) Clonal patterns in phaeochromocytomas and MEN-2A adrenal medullary hyperplasias: Histological and kinetic correlates. J Pathol 192: 221–228. doi: 10.1002/1096-9896(2000)9999:9999<::AID-PATH679>3.0.CO;2-B
|
| [56] | Jones PA (1986) DNA methylation and cancer. Cancer Res 46: 461–466. |
| [57] |
Jones PA, Baylin SB (2007) The epigenomics of cancer. Cell 128: 683–692. doi: 10.1016/j.cell.2007.01.029
|
| [58] |
Diaz-Cano SJ (2001) Are PCR artifacts in microdissected samples preventable? Hum Pathol 32: 1415–1416. doi: 10.1053/hupa.2001.29632
|
| [59] |
Diaz-Cano SJ, Miguel MD, Blanes A, et al. (2001) Contribution of the microvessel network to the clonal and kinetic profiles of adrenal cortical proliferative lesions. Hum Pathol 32: 1232–1239. doi: 10.1053/hupa.2001.28949
|
| [60] |
Zellmer VR, Zhang S (2014) Evolving concepts of tumor heterogeneity. Cell Biosci 4: 69. doi: 10.1186/2045-3701-4-69
|
| [61] |
Blanes A, Rubio J, Sanchez-Carrillo JJ, et al. (2009) Coexistent intraurothelial carcinoma and muscle-invasive urothelial carcinoma of the bladder: Clonality and somatic down-regulation of DNA mismatch repair. Hum Pathol 40: 988–997. doi: 10.1016/j.humpath.2008.12.009
|
| [62] |
Diaz-Cano SJ (2015) Pathological bases for a robust application of cancer molecular classification. Int J Mol Sci 16: 8655–8675. doi: 10.3390/ijms16048655
|
| [63] |
Diaz-Cano SJ, de Miguel M, Blanes A, et al. (2001) Germline RET 634 mutation positive MEN 2A-related C-cell hyperplasias have genetic features consistent with intraepithelial neoplasia. J Clin Endocrinol Metab 86: 3948–3957. doi: 10.1210/jcem.86.8.7739
|
| [64] |
Rubio J, Blanes A, Sanchez-Carrillo JJ, et al. (2007) Microsatellite abnormalities and somatic down-regulation of mismatch repair characterize nodular-trabecular muscle-invasive urothelial carcinoma of the bladder. Histopathology 51: 458–467. doi: 10.1111/j.1365-2559.2007.02795.x
|
| [65] |
Husain EA, Mein C, Pozo L, et al. (2011) Heterogeneous topographic profiles of kinetic and cell cycle regulator microsatellites in atypical (dysplastic) melanocytic nevi. Mod Pathol 24: 471–486. doi: 10.1038/modpathol.2010.143
|
| [66] |
Morrison SJ, Spradling AC (2008) Stem cells and niches: Mechanisms that promote stem cell maintenance throughout life. Cell 132: 598–611. doi: 10.1016/j.cell.2008.01.038
|
| [67] |
Reddig PJ, Juliano RL (2005) Clinging to life: Cell to matrix adhesion and cell survival. Cancer Metastasis Rev 24: 425–439. doi: 10.1007/s10555-005-5134-3
|
| [68] |
Tlsty TD, Coussens LM (2006) Tumor stroma and regulation of cancer development. Annu Rev Pathol 1: 119–150. doi: 10.1146/annurev.pathol.1.110304.100224
|
| [69] |
Baithun SI, Naase M, Blanes A, et al. (2001) Molecular and kinetic features of transitional cell carcinomas of the bladder: Biological and clinical implications. Virchows Arch 438: 289–297. doi: 10.1007/s004280000289
|
| [70] |
Durrett R, Foo J, Leder K, et al. (2011) Intratumor heterogeneity in evolutionary models of tumor progression. Genetics 188: 461–477. doi: 10.1534/genetics.110.125724
|
| [71] |
Laconi E (2007) The evolving concept of tumor microenvironments. Bioessays 29: 738–744. doi: 10.1002/bies.20606
|
| [72] |
Huang LE, Bindra RS, Glazer PM, et al. (2007) Hypoxia-induced genetic instability-a calculated mechanism underlying tumor progression. J Mol Med 85: 139–148. doi: 10.1007/s00109-006-0133-6
|
| [73] |
Hahn WC, Counter CM, Lundberg AS, et al. (1999) Creation of human tumour cells with defined genetic elements. Nature 400: 464–468. doi: 10.1038/22780
|
| [74] |
Rubin H (2003) Microenvironmental regulation of the initiated cell. Adv Cancer Res 90: 1–62. doi: 10.1016/S0065-230X(03)90001-7
|
| [75] |
Mudgil AV, Segal N, Andriani F, et al. (2003) Ultraviolet B irradiation induces expansion of intraepithelial tumor cells in a tissue model of early cancer progression. J Invest Dermatol 121: 191–197. doi: 10.1046/j.1523-1747.2003.12320.x
|
| [76] |
Schollnberger H, Manuguerra M, Bijwaard H, et al. (2006) Analysis of epidemiological cohort data on smoking effects and lung cancer with a multi-stage cancer model. Carcinogenesis 27: 1432–1444. doi: 10.1093/carcin/bgi345
|
| [77] | Farber E, Rubin H (1991) Cellular adaptation in the origin and development of cancer. Cancer Res 51: 2751–2761. |
| [78] |
Cristini V, Frieboes HB, Gatenby R, et al. (2005) Morphologic instability and cancer invasion. Clin Cancer Res 11: 6772–6779. doi: 10.1158/1078-0432.CCR-05-0852
|
| [79] |
Cayama E, Tsuda H, Sarma DS, et al. (1978) Initiation of chemical carcinogenesis requires cell proliferation. Nature 275: 60–62. doi: 10.1038/275060a0
|
| [80] |
Breivik J (2005) The evolutionary origin of genetic instability in cancer development. Semin Cancer Biol 15: 51–60. doi: 10.1016/j.semcancer.2004.09.008
|
| [81] |
West RB, Rubin BP, Miller MA, et al. (2006) A landscape effect in tenosynovial giant-cell tumor from activation of CSF1 expression by a translocation in a minority of tumor cells. Proc Natl Acad Sci U S A 103: 690–695. doi: 10.1073/pnas.0507321103
|
| [82] |
van Vugt MA, Bras A, Medema RH (2005) Restarting the cell cycle when the checkpoint comes to a halt. Cancer Res 65: 7037–7040. doi: 10.1158/0008-5472.CAN-05-1054
|
| [83] |
Jonason AS, Kunala S, Price GJ, et al. (1996) Frequent clones of p53-mutated keratinocytes in normal human skin. Proc Natl Acad Sci U S A 93: 14025–14029. doi: 10.1073/pnas.93.24.14025
|
| [84] |
Laconi E, Pani P, Farber E (2000) The resistance phenotype in the development and treatment of cancer. Lancet Oncol 1: 235–241. doi: 10.1016/S1470-2045(00)00154-6
|
| [85] |
Marongiu F, Doratiotto S, Montisci S, et al. (2008) Liver repopulation and carcinogenesis: Two sides of the same coin? Am J Pathol 172: 857–864. doi: 10.2353/ajpath.2008.070910
|
| [86] |
Campisi J (2007) Aging and cancer cell biology, 2007. Aging Cell 6: 261–263. doi: 10.1111/j.1474-9726.2007.00292.x
|
| [87] |
Sharpless NE, DePinho RA (2007) How stem cells age and why this makes us grow old. Nat Rev Mol Cell Biol 8: 703–713. doi: 10.1038/nrm2241
|
| [88] | McCullough KD, Coleman WB, Smith GJ, et al. (1994) Age-dependent regulation of the tumorigenic potential of neoplastically transformed rat liver epithelial cells by the liver microenvironment. Cancer Res 54: 3668–3671. |
| [89] |
Pasciu D, Montisci S, Greco M, et al. (2006) Aging is associated with increased clonogenic potential in rat liver in vivo. Aging Cell 5: 373–377. doi: 10.1111/j.1474-9726.2006.00230.x
|
| [90] |
Krtolica A, Parrinello S, Lockett S, et al. (2001) Senescent fibroblasts promote epithelial cell growth and tumorigenesis: A link between cancer and aging. Proc Natl Acad Sci U S A 98: 12072–12077. doi: 10.1073/pnas.211053698
|
| [91] |
Liu D, Hornsby PJ (2007) Senescent human fibroblasts increase the early growth of xenograft tumors via matrix metalloproteinase secretion. Cancer Res 67: 3117–3126. doi: 10.1158/0008-5472.CAN-06-3452
|
| [92] | Trost TM, Lausch EU, Fees SA, et al. (2005) Premature senescence is a primary fail-safe mechanism of ERBB2-driven tumorigenesis in breast carcinoma cells. Cancer Res 65: 840–849. |
| [93] |
Philip M, Rowley DA, Schreiber H (2004) Inflammation as a tumor promoter in cancer induction. Semin Cancer Biol 14: 433–439. doi: 10.1016/j.semcancer.2004.06.006
|
| [94] |
Hussain SP, Harris CC (2007) Inflammation and cancer: An ancient link with novel potentials. Int J Cancer 121: 2373–2380. doi: 10.1002/ijc.23173
|
| [95] |
Prehn RT (1997) Regeneration versus neoplastic growth. Carcinogenesis 18: 1439–1444. doi: 10.1093/carcin/18.8.1439
|
| [96] |
Correa P, Houghton J (2007) Carcinogenesis of Helicobacter pylori. Gastroenterology 133: 659–672. doi: 10.1053/j.gastro.2007.06.026
|
| [97] |
De Marzo AM, Marchi VL, Epstein JI, et al. (1999) Proliferative inflammatory atrophy of the prostate: Implications for prostatic carcinogenesis. Am J Pathol 155: 1985–1992. doi: 10.1016/S0002-9440(10)65517-4
|
| [98] |
De Marzo AM, Nakai Y, Nelson WG (2007) Inflammation, atrophy, and prostate carcinogenesis. Urol Oncol 25: 398–400. doi: 10.1016/j.urolonc.2007.05.007
|
| [99] |
Fox JG, Wang TC (2007) Inflammation, atrophy, and gastric cancer. J Clin Invest 117: 60–69. doi: 10.1172/JCI30111
|
| [100] |
Harpaz N (2007) Neoplastic precursor lesions related to the development of cancer in inflammatory bowel disease. Gastroenterol Clin North Am 36: 901–926, vii–viii. doi: 10.1016/j.gtc.2007.08.003
|
| [101] |
Rass U, Ahel I, West SC (2007) Defective DNA repair and neurodegenerative disease. Cell 130: 991–1004. doi: 10.1016/j.cell.2007.08.043
|
| [102] |
De Boer J, Andressoo JO, De Wit J, et al. (2002) Premature aging in mice deficient in DNA repair and transcription. Science 296: 1276–1279. doi: 10.1126/science.1070174
|
| [103] |
Simpson AJ (1997) The natural somatic mutation frequency and human carcinogenesis. Adv Cancer Res 71: 209–240. doi: 10.1016/S0065-230X(08)60100-1
|
| [104] |
Farber E, Cameron R (1980) The sequential analysis of cancer development. Adv Cancer Res 31: 125–226. doi: 10.1016/S0065-230X(08)60658-2
|
| [105] |
Bindra RS, Glazer PM (2005) Genetic instability and the tumor microenvironment: Owards the concept of microenvironment-induced mutagenesis. Mutat Res 569: 75–85. doi: 10.1016/j.mrfmmm.2004.03.013
|
| [106] |
Sneddon JB, Werb Z (2007) Location, location, location: The cancer stem cell niche. Cell Stem Cell 1: 607–611. doi: 10.1016/j.stem.2007.11.009
|
| [107] |
Calabrese C, Poppleton H, Kocak M, et al. (2007) A perivascular niche for brain tumor stem cells. Cancer Cell 11: 69–82. doi: 10.1016/j.ccr.2006.11.020
|
| [108] |
Veeravagu A, Bababeygy SR, Kalani MY, et al. (2008) The cancer stem cell-vascular niche complex in brain tumor formation. Stem Cells Dev 17: 859–867. doi: 10.1089/scd.2008.0047
|
| [109] |
Gilbertson RJ, Gutmann DH (2007) Tumorigenesis in the brain: Location, location, location. Cancer Res 67: 5579–5582. doi: 10.1158/0008-5472.CAN-07-0760
|
| [110] |
Gilbertson RJ, Rich JN (2007) Making a tumour's bed: Glioblastoma stem cells and the vascular niche. Nat Rev Cancer 7: 733–736. doi: 10.1038/nrc2246
|
| [111] |
Croker AK, Goodale D, Chu J, et al. (2009) High aldehyde dehydrogenase and expression of cancer stem cell markers selects for breast cancer cells with enhanced malignant and metastatic ability. J Cell Mol Med 13: 2236–2252. doi: 10.1111/j.1582-4934.2008.00455.x
|
| [112] |
Croker AK, Allan AL (2008) Cancer stem cells: Implications for the progression and treatment of metastatic disease. J Cell Mol Med 12: 374–390. doi: 10.1111/j.1582-4934.2007.00211.x
|
| [113] |
Campbell I, Qiu W, Haviv I (2011) Genetic changes in tumour microenvironments. J Pathol 223: 450–458. doi: 10.1002/path.2842
|
| [114] |
Korkaya H, Liu S, Wicha MS (2011) Breast cancer stem cells, cytokine networks, and the tumor microenvironment. J Clin Invest 121: 3804–3809. doi: 10.1172/JCI57099
|
| [115] |
Weinberg RA (2008) Coevolution in the tumor microenvironment. Nat Genet 40: 494–495. doi: 10.1038/ng0508-494
|
| [116] |
Whiteside TL (2008) The tumor microenvironment and its role in promoting tumor growth. Oncogene 27: 5904–5912. doi: 10.1038/onc.2008.271
|
| [117] |
Ben-Baruch A (2003) Host microenvironment in breast cancer development: Inflammatory cells, cytokines and chemokines in breast cancer progression: Reciprocal tumor-microenvironment interactions. Breast Cancer Res 5: 31–36. doi: 10.1186/bcr690
|
| [118] |
Zipin-Roitman A, Meshel T, Sagi-Assif O, et al. (2007) CXCL10 promotes invasion-related properties in human colorectal carcinoma cells. Cancer Res 67: 3396–3405. doi: 10.1158/0008-5472.CAN-06-3087
|
| [119] |
Malumbres M, Barbacid M (2001) To cycle or not to cycle: A critical decision in cancer. Nat Rev Cancer 1: 222–231. doi: 10.1038/35106065
|
| [120] |
Hersey P, Zhang XD (2001) How melanoma cells evade trail-induced apoptosis. Nat Rev Cancer 1: 142–150. doi: 10.1038/35101078
|
| [121] |
Anderson AR, Hassanein M, Branch KM, et al. (2009) Microenvironmental independence associated with tumor progression. Cancer Res 69: 8797–8806. doi: 10.1158/0008-5472.CAN-09-0437
|
| [122] |
Anderson AR, Rejniak KA, Gerlee P, et al. (2009) Microenvironment driven invasion: A multiscale multimodel investigation. J Math Biol 58: 579–624. doi: 10.1007/s00285-008-0210-2
|
| [123] |
Anderson AR, Weaver AM, Cummings PT, et al. (2006) Tumor morphology and phenotypic evolution driven by selective pressure from the microenvironment. Cell 127: 905–915. doi: 10.1016/j.cell.2006.09.042
|
| [124] |
Coussens LM, Werb Z (2002) Inflammation and cancer. Nature 420: 860–867. doi: 10.1038/nature01322
|
| [125] |
Ellis LM, Hicklin DJ (2008) VEGF-targeted therapy: Mechanisms of anti-tumour activity. Nat Rev Cancer 8: 579–591. doi: 10.1038/nrc2403
|
| [126] |
Yang L, Huang J, Ren X, et al. (2008) Abrogation of TGF beta signaling in mammary carcinomas recruits Gr-1+CD11b+ myeloid cells that promote metastasis. Cancer Cell 13: 23–35. doi: 10.1016/j.ccr.2007.12.004
|
| [127] |
Gabrilovich D (2004) Mechanisms and functional significance of tumour-induced dendritic-cell defects. Nat Rev Immunol 4: 941–952. doi: 10.1038/nri1498
|
| [128] |
Marx J (2008) All in the stroma: Cancer's Cosa Nostra. Science 320: 38–41. doi: 10.1126/science.320.5872.38
|
| [129] |
Marx J (2008) Cancer's bulwark against immune attack: MDS cells. Science 319: 154–156. doi: 10.1126/science.319.5860.154
|
| [130] |
Nonaka K, Saio M, Suwa T, et al. (2008) Skewing the Th cell phenotype toward Th1 alters the maturation of tumor-infiltrating mononuclear phagocytes. J Leukoc Biol 84: 679–688. doi: 10.1189/jlb.1107729
|
| [131] |
Umemura N, Saio M, Suwa T, et al. (2008) Tumor-infiltrating myeloid-derived suppressor cells are pleiotropic-inflamed monocytes/macrophages that bear M1- and M2-type characteristics. J Leukoc Biol 83: 1136–1144. doi: 10.1189/jlb.0907611
|
| [132] |
Mantovani A, Sica A, Allavena P, et al. (2009) Tumor-associated macrophages and the related myeloid-derived suppressor cells as a paradigm of the diversity of macrophage activation. Hum Immunol 70: 325–330. doi: 10.1016/j.humimm.2009.02.008
|
| [133] |
Sica A, Larghi P, Mancino A, et al. (2008) Macrophage polarization in tumour progression. Semin Cancer Biol 18: 349–355. doi: 10.1016/j.semcancer.2008.03.004
|
| [134] |
Mantovani A, Sica A (2010) Macrophages, innate immunity and cancer: Balance, tolerance, and diversity. Curr Opin Immunol 22: 231–237. doi: 10.1016/j.coi.2010.01.009
|
| [135] |
Sinha P, Clements VK, Miller S, et al. (2005) Tumor immunity: A balancing act between T cell activation, macrophage activation and tumor-induced immune suppression. Cancer Immunol Immunother 54: 1137–1142. doi: 10.1007/s00262-005-0703-4
|
| [136] |
Sinha P, Clements VK, Ostrand-Rosenberg S (2005) Reduction of myeloid-derived suppressor cells and induction of M1 macrophages facilitate the rejection of established metastatic disease. J Immunol 174: 636–645. doi: 10.4049/jimmunol.174.2.636
|
| [137] |
Sinha P, Clements VK, Ostrand-Rosenberg S (2005) Interleukin-13-regulated M2 macrophages in combination with myeloid suppressor cells block immune surveillance against metastasis. Cancer Res 65: 11743–11751. doi: 10.1158/0008-5472.CAN-05-0045
|
| [138] |
Lewis CE, Pollard JW (2006) Distinct role of macrophages in different tumor microenvironments. Cancer Res 66: 605–612. doi: 10.1158/0008-5472.CAN-05-4005
|
| [139] |
Yang L, DeBusk LM, Fukuda K, et al. (2004) Expansion of myeloid immune suppressor Gr+CD11b+ cells in tumor-bearing host directly promotes tumor angiogenesis. Cancer Cell 6: 409–421. doi: 10.1016/j.ccr.2004.08.031
|
| [140] |
Sinha P, Clements VK, Bunt SK, et al. (2007) Cross-talk between myeloid-derived suppressor cells and macrophages subverts tumor immunity toward a type 2 response. J Immunol 179: 977–983. doi: 10.4049/jimmunol.179.2.977
|
| [141] |
Condeelis J, Pollard JW (2006) Macrophages: Obligate partners for tumor cell migration, invasion, and metastasis. Cell 124: 263–266. doi: 10.1016/j.cell.2006.01.007
|
| [142] |
Larsen M, Tazzyman S, Lund EL, et al. (2008) Hypoxia-induced secretion of macrophage migration-inhibitory factor from MCF-7 breast cancer cells is regulated in a hypoxia-inducible factor-independent manner. Cancer Lett 265: 239–249. doi: 10.1016/j.canlet.2008.02.012
|
| [143] |
Bosco MC, Puppo M, Blengio F, et al. (2008) Monocytes and dendritic cells in a hypoxic environment: Spotlights on chemotaxis and migration. Immunobiology 213: 733–749. doi: 10.1016/j.imbio.2008.07.031
|
| [144] |
Youn JI, Nagaraj S, Collazo M, et al. (2008) Subsets of myeloid-derived suppressor cells in tumor-bearing mice. J Immunol 181: 5791–5802. doi: 10.4049/jimmunol.181.8.5791
|
| [145] |
Movahedi K, Guilliams M, Van den Bossche J, et al. (2008) Identification of discrete tumor-induced myeloid-derived suppressor cell subpopulations with distinct T cell-suppressive activity. Blood 111: 4233–4244. doi: 10.1182/blood-2007-07-099226
|
| [146] |
Kusmartsev S, Gabrilovich DI (2003) Inhibition of myeloid cell differentiation in cancer: The role of reactive oxygen species. J Leukoc Biol 74: 186–196. doi: 10.1189/jlb.0103010
|
| [147] |
Sinha P, Clements VK, Fulton AM, et al. (2007) Prostaglandin E2 promotes tumor progression by inducing myeloid-derived suppressor cells. Cancer Res 67: 4507–4513. doi: 10.1158/0008-5472.CAN-06-4174
|
| [148] | Bronte V, Serafini P, Mazzoni A, et al. (2003) L-arginine metabolism in myeloid cells controls T-lymphocyte functions. Trends Immunol 24: 302–306. |
| [149] |
Almand B, Clark JI, Nikitina E, et al. (2001) Increased production of immature myeloid cells in cancer patients: A mechanism of immunosuppression in cancer. J Immunol 166: 678–689. doi: 10.4049/jimmunol.166.1.678
|
| [150] |
Baniyash M (2004) TCR zeta-chain downregulation: Curtailing an excessive inflammatory immune response. Nat Rev Immunol 4: 675–687. doi: 10.1038/nri1434
|
| [151] |
Bunt SK, Sinha P, Clements VK, et al. (2006) Inflammation induces myeloid-derived suppressor cells that facilitate tumor progression. J Immunol 176: 284–290. doi: 10.4049/jimmunol.176.1.284
|
| [152] |
Bunt SK, Yang L, Sinha P, et al. (2007) Reduced inflammation in the tumor microenvironment delays the accumulation of myeloid-derived suppressor cells and limits tumor progression. Cancer Res 67: 10019–10026. doi: 10.1158/0008-5472.CAN-07-2354
|
| [153] |
Bielas JH, Loeb LA (2005) Mutator phenotype in cancer: Timing and perspectives. Environ Mol Mutagen 45: 206–213. doi: 10.1002/em.20111
|
| [154] |
Bardelli A, Cahill DP, Lederer G, et al. (2001) Carcinogen-specific induction of genetic instability. Proc Natl Acad Sci U S A 98: 5770–5775. doi: 10.1073/pnas.081082898
|
| [155] |
Jimenez JJ, Blanes A, Diaz-Cano SJ (2003) Microsatellite instability in colon cancer. N Engl J Med 349: 1774–1776. doi: 10.1056/NEJM200310303491818
|
| [156] |
Diaz-Cano SJ (2006) Molecular mechanisms in melanoma. N Engl J Med 355: 1395. doi: 10.1056/NEJMc062093
|
| [157] |
Pozo L, Husein E, Blanes A, et al. (2008) The correlation of regression with the grade of dysplasia (atypia) in melanocytic naevi. Histopathology 52: 387–389. doi: 10.1111/j.1365-2559.2007.02880.x
|
| [158] |
Husemann Y, Geigl JB, Schubert F, et al. (2008) Systemic spread is an early step in breast cancer. Cancer Cell 13: 58–68. doi: 10.1016/j.ccr.2007.12.003
|
| [159] |
Arif S, Patel J, Blanes A, et al. (2007) Cytoarchitectural and kinetic features in the histological evaluation of follicular thyroid neoplasms. Histopathology 50: 750–763. doi: 10.1111/j.1365-2559.2007.02680.x
|
| [160] |
Boone CW, Kelloff GJ, Freedman LS (1993) Intraepithelial and postinvasive neoplasia as a stochastic continuum of clonal evolution, and its relationship to mechanisms of chemopreventive drug action. J Cell Biochem Suppl 53: 14–25. doi: 10.1002/jcb.240531104
|
| [161] |
Namba R, Maglione JE, Davis RR, et al. (2006) Heterogeneity of mammary lesions represent molecular differences. BMC Cancer 6: 275. doi: 10.1186/1471-2407-6-275
|
| [162] |
Steeg PS (2006) Tumor metastasis: Mechanistic insights and clinical challenges. Nat Med 12: 895–904. doi: 10.1038/nm1469
|
| [163] |
Steeg PS (2007) Cancer: Icromanagement of metastasis. Nature 449: 671–673. doi: 10.1038/449671a
|
| [164] |
Talmadge JE (2007) Clonal selection of metastasis within the life history of a tumor. Cancer Res 67: 11471–11475. doi: 10.1158/0008-5472.CAN-07-2496
|
| [165] |
Christofori G (2006) New signals from the invasive front. Nature 441: 444–450. doi: 10.1038/nature04872
|
| [166] |
Gupta GP, Massague J (2006) Cancer metastasis: Building a framework. Cell 127: 679–695. doi: 10.1016/j.cell.2006.11.001
|
| [167] | Nguyen DX (2011) Tracing the origins of metastasis. J Pathol 223: 195–204. |
| [168] | Nguyen DX, Massague J (2007) Genetic determinants of cancer metastasis. Nat Rev Genet 8: 341–352. |
| [169] |
Li F, Tiede B, Massague J, et al. (2007) Beyond tumorigenesis: Cancer stem cells in metastasis. Cell Res 17: 3–14. doi: 10.1038/sj.cr.7310118
|
| [170] |
Wicha MS (2006) Cancer stem cells and metastasis: Lethal seeds. Clin Cancer Res 12: 5606–5607. doi: 10.1158/1078-0432.CCR-06-1537
|
| [171] | Kaplan RN, Psaila B, Lyden D (2006) Bone marrow cells in the 'pre-metastatic niche': Within bone and beyond. Cancer Metastasis Rev 25: 521–529. |
| [172] |
Kaplan RN, Rafii S, Lyden D (2006) Preparing the "soil": The premetastatic niche. Cancer Res 66: 11089–11093. doi: 10.1158/0008-5472.CAN-06-2407
|
| [173] |
Kaplan RN, Riba RD, Zacharoulis S, et al. (2005) VEGFR1-positive haematopoietic bone marrow progenitors initiate the pre-metastatic niche. Nature 438: 820–827. doi: 10.1038/nature04186
|
| [174] |
Bissig H, Richter J, Desper R, et al. (1999) Evaluation of the clonal relationship between primary and metastatic renal cell carcinoma by comparative genomic hybridization. Am J Pathol 155: 267–274. doi: 10.1016/S0002-9440(10)65120-6
|
| [175] |
Cheng L, Bostwick DG, Li G, et al. (1999) Allelic imbalance in the clonal evolution of prostate carcinoma. Cancer 85: 2017–2022. doi: 10.1002/(SICI)1097-0142(19990501)85:9%3C2017::AID-CNCR20%3E3.0.CO;2-V
|
| [176] | Kuukasjarvi T, Karhu R, Tanner M, et al. (1997) Genetic heterogeneity and clonal evolution underlying development of asynchronous metastasis in human breast cancer. Cancer Res 57: 1597–1604. |
| [177] | Ramaswamy S, Ross KN, Lander ES, et al. (2003) A molecular signature of metastasis in primary solid tumors. Nat Genet 33: 49–54. |
| [178] |
Weigelt B, Glas AM, Wessels LF, et al. (2003) Gene expression profiles of primary breast tumors maintained in distant metastases. Proc Natl Acad Sci U S A 100: 15901–15905. doi: 10.1073/pnas.2634067100
|
| [179] |
Weigelt B, Hu Z, He X, et al. (2005) Molecular portraits and 70-gene prognosis signature are preserved throughout the metastatic process of breast cancer. Cancer Res 65: 9155–9158. doi: 10.1158/0008-5472.CAN-05-2553
|
| [180] |
Liu W, Laitinen S, Khan S, et al. (2009) Copy number analysis indicates monoclonal origin of lethal metastatic prostate cancer. Nat Med 15: 559–565. doi: 10.1038/nm.1944
|
| [181] | Ruijter ET, van de Kaa CA, Schalken JA, et al. (1996) Histological grade heterogeneity in multifocal prostate cancer. Biological and clinical implications. J Pathol 180: 295–299. |
| [182] |
Alvarado C, Beitel LK, Sircar K, et al. (2005) Somatic mosaicism and cancer: A micro-genetic examination into the role of the androgen receptor gene in prostate cancer. Cancer Res 65: 8514–8518. doi: 10.1158/0008-5472.CAN-05-0399
|
| [183] |
Shah SP, Morin RD, Khattra J, et al. (2009) Mutational evolution in a lobular breast tumour profiled at single nucleotide resolution. Nature 461: 809–813. doi: 10.1038/nature08489
|
| [184] |
Holmgren L, O'Reilly MS, Folkman J (1995) Dormancy of micrometastases: Balanced proliferation and apoptosis in the presence of angiogenesis suppression. Nat Med 1: 149–153. doi: 10.1038/nm0295-149
|
| [185] |
Braun S, Pantel K, Muller P, et al. (2000) Cytokeratin-positive cells in the bone marrow and survival of patients with stage I, II, or III breast cancer. N Engl J Med 342: 525–533. doi: 10.1056/NEJM200002243420801
|
| [186] |
Diaz-Cano SJ (2000) Bone marrow metastases in breast cancer. N Engl J Med 343: 577. doi: 10.1056/NEJM200008243430811
|
| [187] |
Passlick B, Izbicki JR, Kubuschok B, et al. (1994) Immunohistochemical assessment of individual tumor cells in lymph nodes of patients with non-small-cell lung cancer. J Clin Oncol 12: 1827–1832. doi: 10.1200/JCO.1994.12.9.1827
|
| [188] |
Schlimok G, Funke I, Holzmann B, et al. (1987) Micrometastatic cancer cells in bone marrow: In vitro detection with anti-cytokeratin and in vivo labeling with anti-17-1A monoclonal antibodies. Proc Natl Acad Sci U S A 84: 8672–8676. doi: 10.1073/pnas.84.23.8672
|
| [189] |
Smith BL (2000) Approaches to breast-cancer staging. N Engl J Med 342: 580–581. doi: 10.1056/NEJM200002243420809
|
| [190] |
Klein CA, Schmidt-Kittler O, Schardt JA, et al. (1999) Comparative genomic hybridization, loss of heterozygosity, and DNA sequence analysis of single cells. Proc Natl Acad Sci U S A 96: 4494–4499. doi: 10.1073/pnas.96.8.4494
|
| [191] |
Klein CA, Blankenstein TJ, Schmidt-Kittler O, et al. (2002) Genetic heterogeneity of single disseminated tumour cells in minimal residual cancer. Lancet 360: 683–689. doi: 10.1016/S0140-6736(02)09838-0
|
| [192] |
Schmidt-Kittler O, Ragg T, Daskalakis A, et al. (2003) From latent disseminated cells to overt metastasis: Genetic analysis of systemic breast cancer progression. Proc Natl Acad Sci U S A 100: 7737–7742. doi: 10.1073/pnas.1331931100
|
| [193] |
Bedenne L, Michel P, Bouche O, et al. (2007) Chemoradiation followed by surgery compared with chemoradiation alone in squamous cancer of the esophagus: FFCD 9102. J Clin Oncol 25: 1160–1168. doi: 10.1200/JCO.2005.04.7118
|
| [194] |
Stoecklein NH, Hosch SB, Bezler M, et al. (2008) Direct genetic analysis of single disseminated cancer cells for prediction of outcome and therapy selection in esophageal cancer. Cancer Cell 13: 441–453. doi: 10.1016/j.ccr.2008.04.005
|
| [195] |
Podsypanina K, Du YC, Jechlinger M, et al. (2008) Seeding and propagation of untransformed mouse mammary cells in the lung. Science 321: 1841–1844. doi: 10.1126/science.1161621
|
| [196] |
Gadi VK, Nelson JL (2007) Fetal microchimerism in women with breast cancer. Cancer Res 67: 9035–9038. doi: 10.1158/0008-5472.CAN-06-4209
|
| [197] |
Solakoglu O, Maierhofer C, Lahr G, et al. (2002) Heterogeneous proliferative potential of occult metastatic cells in bone marrow of patients with solid epithelial tumors. Proc Natl Acad Sci U S A 99: 2246–2251. doi: 10.1073/pnas.042372199
|
| [198] |
Gangnus R, Langer S, Breit E, et al. (2004) Genomic profiling of viable and proliferative micrometastatic cells from early-stage breast cancer patients. Clin Cancer Res 10: 3457–3464. doi: 10.1158/1078-0432.CCR-03-0818
|
| [199] |
Abbruzzese JL, Abbruzzese MC, Hess KR, et al. (1994) Unknown primary carcinoma: Natural history and prognostic factors in 657 consecutive patients. J Clin Oncol 12: 1272–1280. doi: 10.1200/JCO.1994.12.6.1272
|
| [200] |
Klein CA, Holzel D (2006) Systemic cancer progression and tumor dormancy: Mathematical models meet single cell genomics. Cell Cycle 5: 1788–1798. doi: 10.4161/cc.5.16.3097
|
| [201] |
Beerenwinkel N, Antal T, Dingli D, et al. (2007) Genetic progression and the waiting time to cancer. PLoS Comput Biol 3: 225. doi: 10.1371/journal.pcbi.0030225
|
| [202] |
Sieber OM, Tomlinson SR, Tomlinson IP (2005) Tissue, cell and stage specificity of (epi)mutations in cancers. Nat Rev Cancer 5: 649–655. doi: 10.1038/nrc1674
|
| [203] |
Fialkow PJ (1979) Clonal origin of human tumors. Annu Rev Med 30: 135–143. doi: 10.1146/annurev.me.30.020179.001031
|
| [204] | Weinberg RA (2007) The biology of cancer, New York: Garland Science, 796. |
| [205] |
Novelli MR, Williamson JA, Tomlinson IP, et al. (1996) Polyclonal origin of colonic adenomas in an XO/XY patient with FAP. Science 272: 1187–1190. doi: 10.1126/science.272.5265.1187
|
| [206] |
Leroi AM, Koufopanou V, Burt A (2003) Cancer selection. Nat Rev Cancer 3: 226–231. doi: 10.1038/nrc1016
|
| [207] |
Potten CS (1998) Stem cells in gastrointestinal epithelium: Numbers, characteristics and death. Philos Trans R Soc Lond 353: 821–830. doi: 10.1098/rstb.1998.0246
|
| [208] |
Friedberg EC (2003) DNA damage and repair. Nature 421: 436–440. doi: 10.1038/nature01408
|
| [209] |
Tomlinson I, Bodmer W (1999) Selection, the mutation rate and cancer: Ensuring that the tail does not wag the dog. Nat Med 5: 11–12. doi: 10.1038/4687
|
| [210] |
Wang TL, Rago C, Silliman N, et al. (2002) Prevalence of somatic alterations in the colorectal cancer cell genome. Proc Natl Acad Sci U S A 99: 3076–3080. doi: 10.1073/pnas.261714699
|
| [211] |
Bielas JH, Loeb KR, Rubin BP, et al. (2006) Human cancers express a mutator phenotype. Proc Natl Acad Sci U S A 103: 18238–18242. doi: 10.1073/pnas.0607057103
|
| [212] |
Loeb LA (2016) Human cancers express a mutator phenotype: Hypothesis, origin, and consequences. Cancer Res 76: 2057–2059. doi: 10.1158/0008-5472.CAN-16-0794
|
| [213] |
Diaz-Cano SJ (2007) Kinetic topographical heterogeneity in follicular thyroid neoplasms and growth patterns. Histopathology 51: 416–418. doi: 10.1111/j.1365-2559.2007.02778.x
|
| [214] |
Gonzalez-Garcia I, Sole RV, Costa J (2002) Metapopulation dynamics and spatial heterogeneity in cancer. Proc Natl Acad Sci U S A 99: 13085–13089. doi: 10.1073/pnas.202139299
|
| [215] |
Gatenby RA, Silva AS, Gillies RJ, et al. (2009) Adaptive therapy. Cancer Res 69: 4894–4903. doi: 10.1158/0008-5472.CAN-08-3658
|
| [216] | Gatenby RA, Vincent TL (2003) An evolutionary model of carcinogenesis. Cancer Res 63: 6212–6220. |
| [217] |
Chan DA, Giaccia AJ (2007) Hypoxia, gene expression, and metastasis. Cancer Metastasis Rev 26: 333–339. doi: 10.1007/s10555-007-9063-1
|
| [218] |
Evans SM, Hahn SM, Magarelli DP, et al. (2001) Hypoxic heterogeneity in human tumors: EF5 binding, vasculature, necrosis, and proliferation. Am J Clin Oncol 24: 467–472. doi: 10.1097/00000421-200110000-00011
|
| [219] |
Martinive P, Defresne F, Quaghebeur E, et al. (2009) Impact of cyclic hypoxia on HIF-1alpha regulation in endothelial cells-new insights for anti-tumor treatments. FEBS J 276: 509–518. doi: 10.1111/j.1742-4658.2008.06798.x
|
| [220] |
Kim Y, Lin Q, Glazer PM, et al. (2009) Hypoxic tumor microenvironment and cancer cell differentiation. Curr Mol Med 9: 425–434. doi: 10.2174/156652409788167113
|
| [221] |
Harris AL (2002) Hypoxia-a key regulatory factor in tumour growth. Nat Rev Cancer 2: 38–47. doi: 10.1038/nrc704
|
| [222] |
Subarsky P, Hill RP (2003) The hypoxic tumour microenvironment and metastatic progression. Clin Exp Metastasis 20: 237–250. doi: 10.1023/A:1022939318102
|
| [223] |
Pouyssegur J, Dayan F, Mazure NM (2006) Hypoxia signalling in cancer and approaches to enforce tumour regression. Nature 441: 437–443. doi: 10.1038/nature04871
|
| [224] |
Semenza GL (2003) Targeting HIF-1 for cancer therapy. Nat Rev Cancer 3: 721–732. doi: 10.1038/nrc1187
|
| [225] | Struckmann K, Mertz K, Steu S, et al. (2008) pVHL co-ordinately regulates CXCR4/CXCL12 and MMP2/MMP9 expression in human clear-cell renal cell carcinoma. J Pathol 214: 464–471. |
| [226] |
Keith B, Simon MC (2007) Hypoxia-inducible factors, stem cells, and cancer. Cell 129: 465–472. doi: 10.1016/j.cell.2007.04.019
|
| [227] |
Bos R, Zhong H, Hanrahan CF, et al. (2001) Levels of hypoxia-inducible factor-1 alpha during breast carcinogenesis. J Natl Cancer Inst 93: 309–314. doi: 10.1093/jnci/93.4.309
|
| [228] |
Semenza GL (2003) Angiogenesis in ischemic and neoplastic disorders. Annu Rev Med 54: 17–28. doi: 10.1146/annurev.med.54.101601.152418
|
| [229] |
Serrati S, Margheri F, Fibbi G, et al. (2008) Endothelial cells and normal breast epithelial cells enhance invasion of breast carcinoma cells by CXCR-4-dependent up-regulation of urokinase-type plasminogen activator receptor (uPAR, CD87) expression. J Pathol 214: 545–554. doi: 10.1002/path.2309
|
| [230] | Jankowski K, Kucia M, Wysoczynski M, et al. (2003) Both hepatocyte growth factor (HGF) and stromal-derived factor-1 regulate the metastatic behavior of human rhabdomyosarcoma cells, but only HGF enhances their resistance to radiochemotherapy. Cancer Res 63: 7926–7935. |
| [231] |
Libura J, Drukala J, Majka M, et al. (2002) CXCR4-SDF-1 signaling is active in rhabdomyosarcoma cells and regulates locomotion, chemotaxis, and adhesion. Blood 100: 2597–2606. doi: 10.1182/blood-2002-01-0031
|
| [232] |
Maeda S, Shinchi H, Kurahara H, et al. (2008) CD133 expression is correlated with lymph node metastasis and vascular endothelial growth factor-C expression in pancreatic cancer. Br J Cancer 98: 1389–1397. doi: 10.1038/sj.bjc.6604307
|
| [233] |
Brabletz T, Jung A, Spaderna S, et al. (2005) Opinion: Migrating cancer stem cells-an integrated concept of malignant tumour progression. Nat Rev Cancer 5: 744–749. doi: 10.1038/nrc1694
|
| [234] |
Kang Y, Massague J (2004) Epithelial-mesenchymal transitions: Twist in development and metastasis. Cell 118: 277–279. doi: 10.1016/j.cell.2004.07.011
|
| [235] |
Pomerance M, Quillard J, Chantoux F, et al. (2006) High-level expression, activation, and subcellular localization of p38-MAP kinase in thyroid neoplasms. J Pathol 209: 298–306. doi: 10.1002/path.1975
|
| [236] |
Murphy PM (2001) Chemokines and the molecular basis of cancer metastasis. N Engl J Med 345: 833–835. doi: 10.1056/NEJM200109133451113
|
| [237] |
Stainier D (2006) No stem cell is an islet (yet). N Engl J Med 354: 521–523. doi: 10.1056/NEJMcibr055048
|
| [238] |
Zlotnik A (2008) New insights on the role of CXCR4 in cancer metastasis. J Pathol 215: 211–213. doi: 10.1002/path.2350
|
| [239] |
DeNardo DG, Johansson M, Coussens LM (2008) Immune cells as mediators of solid tumor metastasis. Cancer Metastasis Rev 27: 11–18. doi: 10.1007/s10555-007-9100-0
|
| [240] |
Hiratsuka S, Watanabe A, Sakurai Y, et al. (2008) The S100A8-serum amyloid A3-TLR4 paracrine cascade establishes a pre-metastatic phase. Nat Cell Biol 10: 1349–1355. doi: 10.1038/ncb1794
|
| [241] |
Bornstein SR, Hornsby PJ (2005) What can we learn from gene expression profiling for adrenal tumor management? J Clin Endocrinol Metab 90: 1900–1902. doi: 10.1210/jc.2005-0065
|
| [242] |
Garber ME, Troyanskaya OG, Schluens K, et al. (2001) Diversity of gene expression in adenocarcinoma of the lung. Proc Natl Acad Sci U S A 98: 13784–13789. doi: 10.1073/pnas.241500798
|
| [243] |
Steeg PS (2005) New insights into the tumor metastatic process revealed by gene expression profiling. Am J Pathol 166: 1291–1294. doi: 10.1016/S0002-9440(10)62348-6
|
| [244] |
van de Rijn M, Rubin BP (2002) Gene expression studies on soft tissue tumors. Am J Pathol 161: 1531–1534. doi: 10.1016/S0002-9440(10)64430-6
|
| [245] |
van't Veer LJ, Paik S, Hayes DF (2005) Gene expression profiling of breast cancer: A new tumor marker. J Clin Oncol 23: 1631–1635. doi: 10.1200/JCO.2005.12.005
|
| [246] | Vaupel P, Harrison L (2004) Tumor hypoxia: Causative factors, compensatory mechanisms, and cellular response. Oncologist 5: 4–9. |
| [247] |
Pennacchietti S, Michieli P, Galluzzo M, et al. (2003) Hypoxia promotes invasive growth by transcriptional activation of the met protooncogene. Cancer Cell 3: 347–361. doi: 10.1016/S1535-6108(03)00085-0
|
| [248] |
Kulshreshtha R, Davuluri RV, Calin GA, et al. (2008) A microRNA component of the hypoxic response. Cell Death Differ 15: 667–671. doi: 10.1038/sj.cdd.4402310
|
| [249] |
Ouatas T, Salerno M, Palmieri D, et al. (2003) Basic and translational advances in cancer metastasis: Nm23. J Bioenerg Biomembr 35: 73–79. doi: 10.1023/A:1023497924277
|
| [250] |
Salerno M, Ouatas T, Palmieri D, et al. (2003) Inhibition of signal transduction by the nm23 metastasis suppressor: Possible mechanisms. Clin Exp Metastasis 20: 3–10. doi: 10.1023/A:1022578000022
|
| [251] |
Steeg PS (2003) Metastasis suppressors alter the signal transduction of cancer cells. Nat Rev Cancer 3: 55–63. doi: 10.1038/nrc967
|
| [252] |
Steeg PS, Ouatas T, Halverson D, et al. (2003) Metastasis suppressor genes: Basic biology and potential clinical use. Clin Breast Cancer 4: 51–62. doi: 10.3816/CBC.2003.n.012
|
| [253] |
Wulfkuhle JD, Paweletz CP, Steeg PS, et al. (2003) Proteomic approaches to the diagnosis, treatment, and monitoring of cancer. Adv Exp Med Biol 532: 59–68. doi: 10.1007/978-1-4615-0081-0_7
|
| [254] |
Guo Y, Feng Y, Trivedi NS, et al. (2011) Medusa structure of the gene regulatory network: Dominance of transcription factors in cancer subtype classification. Exp Biol Med 236: 628–636. doi: 10.1258/ebm.2011.010324
|
| [255] |
Guo Z, Wu F, Asplund A, et al. (2001) Analysis of intratumoral heterogeneity of chromosome 3p deletions and genetic evidence of polyclonal origin of cervical squamous carcinoma. Mod Pathol 14: 54–61. doi: 10.1038/modpathol.3880256
|
| [256] |
Nakamura T, Kuwai T, Kitadai Y, et al. (2007) Zonal heterogeneity for gene expression in human pancreatic carcinoma. Cancer Res 67: 7597–7604. doi: 10.1158/0008-5472.CAN-07-0874
|
| [257] |
Shipitsin M, Campbell LL, Argani P, et al. (2007) Molecular definition of breast tumor heterogeneity. Cancer Cell 11: 259–273. doi: 10.1016/j.ccr.2007.01.013
|
| [258] |
Edelman EJ, Guinney J, Chi JT, et al. (2008) Modeling cancer progression via pathway dependencies. PLoS Comput Biol 4: e28. doi: 10.1371/journal.pcbi.0040028
|
| [259] | Califano J, van der Riet P, Westra W, et al. (1996) Genetic progression model for head and neck cancer: Implications for field cancerization. Cancer Res 56: 2488–2492. |
| [260] |
Parsons DW, Jones S, Zhang X, et al. (2008) An integrated genomic analysis of human glioblastoma multiforme. Science 321: 1807–1812. doi: 10.1126/science.1164382
|
| [261] |
Wood LD, Parsons DW, Jones S, et al. (2007) The genomic landscapes of human breast and colorectal cancers. Science 318: 1108–1113. doi: 10.1126/science.1145720
|
| [262] |
Siegmund KD, Marjoram P, Woo YJ, et al. (2009) Inferring clonal expansion and cancer stem cell dynamics from DNA methylation patterns in colorectal cancers. Proc Natl Acad Sci U S A 106: 4828–4833. doi: 10.1073/pnas.0810276106
|
| [263] |
Campbell PJ, Pleasance ED, Stephens PJ, et al. (2008) Subclonal phylogenetic structures in cancer revealed by ultra-deep sequencing. Proc Natl Acad Sci U S A 105: 13081–13086. doi: 10.1073/pnas.0801523105
|
| [264] |
Mullighan CG, Phillips LA, Su X, et al. (2008) Genomic analysis of the clonal origins of relapsed acute lymphoblastic leukemia. Science 322: 1377–1380. doi: 10.1126/science.1164266
|
| [265] |
Panzer-Grumayer ER, Cazzaniga G, van der Velden VH, et al. (2005) Immunogenotype changes prevail in relapses of young children with TEL-AML1-positive acute lymphoblastic leukemia and derive mainly from clonal selection. Clin Cancer Res 11: 7720–7727. doi: 10.1158/1078-0432.CCR-05-1239
|
| [266] |
Tomlinson IP, Lambros MB, Roylance RR (2002) Loss of heterozygosity analysis: Practically and conceptually flawed? Genes Chromosomes Cancer 34: 349–353. doi: 10.1002/gcc.10085
|
| [267] |
Park SY, Gonen M, Kim HJ, et al. (2010) Cellular and genetic diversity in the progression of in situ human breast carcinomas to an invasive phenotype. J Clin Invest 120: 636–644. doi: 10.1172/JCI40724
|
| [268] |
Diaz-Cano S (1996) PCR-based alternative for diagnosis of immunoglobulin heavy chain gene rearrangement: Principles, practice, and polemics. Diagn Mol Pathol 5: 3–9. doi: 10.1097/00019606-199603000-00002
|
| [269] |
Brady SP, Magro CM, Diaz-Cano SJ, et al. (1999) Analysis of clonality of atypical cutaneous lymphoid infiltrates associated with drug therapy by PCR/DGGE. Hum Pathol 30: 130–136. doi: 10.1016/S0046-8177(99)90266-6
|
| [270] | Teixeira MR, Pandis N, Bardi G, et al. (1996) Karyotypic comparisons of multiple tumorous and macroscopically normal surrounding tissue samples from patients with breast cancer. Cancer Res 56: 855–859. |
| [271] |
Teixeira MR, Pandis N, Bardi G, et al. (1995) Clonal heterogeneity in breast cancer: Karyotypic comparisons of multiple intra- and extra-tumorous samples from 3 patients. Int J Cancer 63: 63–68. doi: 10.1002/ijc.2910630113
|
| [272] | Giaretti W, Monaco R, Pujic N, et al. (1996) Intratumor heterogeneity of K-ras2 mutations in colorectal adenocarcinomas: Association with degree of DNA aneuploidy. Am J Pathol 149: 237–245. |
| [273] | Coons SW, Johnson PC, Shapiro JR (1995) Cytogenetic and flow cytometry DNA analysis of regional heterogeneity in a low grade human glioma. Cancer Res 55: 1569–1577. |
| [274] |
Kallioniemi OP (1988) Comparison of fresh and paraffin-embedded tissue as starting material for DNA flow cytometry and evaluation of intratumor heterogeneity. Cytometry 9: 164–169. doi: 10.1002/cyto.990090211
|
| [275] |
Lage JM, Leamon JH, Pejovic T, et al. (2003) Whole genome analysis of genetic alterations in small DNA samples using hyperbranched strand displacement amplification and array-CGH. Genome Res 13: 294–307. doi: 10.1101/gr.377203
|
| [276] | Leith JT, Michelson S, Faulkner LE, et al. (1987) Growth properties of artificial heterogeneous human colon tumors. Cancer Res 47: 1045–1051. |
| [277] |
Farber L, Efrati E, Elkin H, et al. (2011) Molecular morphometric analysis shows relative intra-tumoural homogeneity for KRAS mutations in colorectal cancer. Virchows Arch 459: 487–493. doi: 10.1007/s00428-011-1158-y
|
| [278] |
Kuwai T, Nakamura T, Kim SJ, et al. (2008) Intratumoral heterogeneity for expression of tyrosine kinase growth factor receptors in human colon cancer surgical specimens and orthotopic tumors. Am J Pathol 172: 358–366. doi: 10.2353/ajpath.2008.070625
|
| [279] |
Inda MM, Bonavia R, Mukasa A, et al. (2010) Tumor heterogeneity is an active process maintained by a mutant EGFR-induced cytokine circuit in glioblastoma. Genes Dev 24: 1731–1745. doi: 10.1101/gad.1890510
|
| [280] | Konishi N, Hiasa Y, Matsuda H, et al. (1995) Intratumor cellular heterogeneity and alterations in ras oncogene and p53 tumor suppressor gene in human prostate carcinoma. Am J Pathol 147: 1112–1122. |
| [281] | Maley CC, Reid BJ, Forrest S (2004) Cancer prevention strategies that address the evolutionary dynamics of neoplastic cells: Simulating benign cell boosters and selection for chemosensitivity. Cancer Epidemiol Biomarkers Prev 13: 1375–1384. |
| [282] | Cottu PH, Asselah J, Lae M, et al. (2008) Intratumoral heterogeneity of HER2/neu expression and its consequences for the management of advanced breast cancer. Ann Oncol 19: 595–597. |

Fatima Al-Hashimi, Salvador J. Diaz-Cano. Multi-target analysis of neoplasms for the evaluation of tumor progression: stochastic approach of biologic processes[J]. AIMS Molecular Science, 2018, 5(1): 14-62. doi: 10.3934/molsci.2018.1.14

 DownLoad:
DownLoad: 


