Citation: Shuyi Cen, Kaiyou Fu, Yue Shi, Hanliang Jiang, Jiawei Shou, Liangkun You, Weidong Han, Hongming Pan, Zhen Liu. A microRNA disease signature associated with lymph node metastasis of lung adenocarcinoma[J]. Mathematical Biosciences and Engineering, 2020, 17(3): 2557-2568. doi: 10.3934/mbe.2020140

| [1] | K. X. Sun, R. S. Zheng, H. M. Zeng, S. W. Zhang, X. N. Zou, X. Y. Gu, et al., The incidence and mortality of lung cancer in China, 2014, Zhonghua Zhong Liu Za Zhi, 40 (2018), 805-811. |
| [2] | R. L. Siegel, K. D. Miller, A. Jemal, Cancer statistics, 2018, CA Cancer J. Clin., 68 (2018), 7-30. |
| [3] | A. McIntyre, A. K. Ganti, Lung cancer-a global perspective, J. Surg. Oncol, 115 (2017), 550-554. |
| [4] | A. Jemal, M. M. Center, C. DeSantis, E. M. Ward, Global patterns of cancer incidence and mortality rates and trends, Cancer Epidemiol. Biomarkers Prev., 19 (2010), 1893-1907. |
| [5] | M. J. Duffy, Clinical uses of tumor markers: A critical review, Crit. Rev. Clin. Lab. Sci., 38 (2001), 225-262. |
| [6] | P. Goldstraw, K. Chansky, J. Crowley, R. R. Porta, H. Asamura, W. E. Eberhardt, et al., The IASLC lung cancer staging project: Proposals for revision of the TNM stage groupings in the forthcoming (eighth) edition of the TNM classification for lung cancer, J. Thorac. Oncol, 11 (2016), 39-51. |
| [7] | A. Matsuda, K. Katanoda, Five-year relative survival rate of lung cancer in the USA, Europe and Japan, Jpn J. Clin. Oncol, 43 (2013), 1287-1288. |
| [8] | Y. Cai, X. Yu, S. Hu, J. Yu, A brief review on the mechanisms of miRNA regulation, Genomics, Proteomics Bioinf., 7 (2009), 147-154. |
| [9] | E. Chan, D. E. Prado, J. B. Weidhaas, Cancer microRNAs: From subtype profiling to predictors of response to therapy, Trends Mol. Med., 17 (2011), 235-243. |
| [10] | J. Ma, K. Mannoor, L. Gao, A. Tan, M. A. Guarnera, M. Zhan, et al., Characterization of microRNA transcriptome in lung cancer by next-generation deep sequencing, Mol. Oncol, 8 (2014), 1208-1219. |
| [11] | A. E. Kerscher, F. J. Slack, Oncomirs-microRNAs with a role in cancer, Nat. Rev. Cancer, 6 (2006), 259-269. |
| [12] | C. Sanfiorenzo, M. I. Ilie, A. Belaid, F. Barlesi, J. Mouroux, C. H. Marquette, et al., Two panels of plasma microRNAs as non-invasive biomarkers for prediction of recurrence in resectable NSCLC, PLoS One, 8 (2013), e54596. |
| [13] | J. Y. Kwan, P. Psarianos, J. P. Bruce, K. W. Yip, F. F. Liu, The complexity of microRNAs in human cancer, J. Radiat. Res., 57 (2016), i106-i111. |
| [14] | R. Tibshirani, Regression shrinkage and selection via the lasso, J. Roy. Stat. Soc. B, 58 (1996), 267-288. |
| [15] | C. Chen, H. Chen, Y. He, R Xia, TBtools, a toolkit for biologists integrating various HTS-data handling tools with a user-friendly interface, Bio. Rxiv, (2018), 289660. |
| [16] | H. Dweep, N. Gretz, miRWalk2. 0: A comprehensive atlas of microRNA-target interactions, Nat. Methods, 12 (2015), 697. |
| [17] | P. Shannon, A. Markiel, O. Ozier, N. S. Baliga, J. T. Wang, D. Ramage, et al., Cytoscape: A software environment for integrated models of biomolecular interaction networks, Genome Res., 13 (2003), 2498-2504. |
| [18] | F. Yang, K. Wei, Z. Qin, W. Liu, C, Shao, C. Wang, et al., MiR-598 suppresses invasion and migration by negative regulation of derlin-1 and epithelial-mesenchymal transition in non-small cell lung cancer, Cell Physiol. Biochem., 47 (2018), 245-256. |
| [19] | H. Y. Lee, S. S. Han, S. Y. Song, Serum microRNAs as potential biomarkers for lung cancer, Ann. Oncol., 27 (2016). |
| [20] | X. H. Wang, Y. Lu, J. J. Liang, J. X. Cao, Y. Q. Jin, G. S. An, et al., MiR-509-3-5p causes aberrant mitosis and anti-proliferative effect by suppression of PLK1 in human lung cancer A549 cells, Biochem. Biophy. Res. Commun., 478 (2016), 676-682. |
| [21] | Y. Z. Wang, J. M. Li, H. M. Chen, Y. Mo, H. Ye, Y. Luo, et al., Down-regulation of miR-133a as a poor prognosticator in non-small cell lung cancer, Gene, 591 (2016), 333-337. |
| [22] | F. Bray, J. Ferlay, I. Soerjomataram, L. A. Torre, A. Jemal, Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries, CA Cancer J. Clin., 68 (2018), 394-424. |
| [23] | M. Cao, W. Chen, Epidemiology of lung cancer in China, Thorac. Cancer, 10 (2019), 3-7. |
| [24] | F. R. Vamos, J. Tovari, J. Fillinger, J. Timar, S. Paku, I. Kenessey, et al., Lymphangiogenesis correlates with lymph node metastasis, prognosis, and angiogenic phenotype in human non-small cell lung cancer, Clin. Cancer Res., 11 (2005), 7344-7353. |
| [25] | S. L. Yu, H. Y. Chen, G. C. Chang, et al., MicroRNA signature predicts survival and relapse in lung cancer, Cancer Cell, 13 (2008), 48-57. |
| [26] | S. Y. Sathipati, S. Y. Ho, Identifying the miRNA signature associated with survival time in patients with lung adenocarcinoma using miRNA expression profiles, Sci. Rep., 7 (2017). |
| [27] | L. Fang, J. Cai, B. Chen, S. Wu, R, Li, X. Xu, et al., Aberrantly expressed miR-582-3p maintains lung cancer stem cell-like traits by activating Wnt/β-catenin signalling, Nat. Commun., 6 (2015), 8640. |
| [28] | J. Liu, S. Liu, X. Deng, J. Rao, K. Huang, G. Xu, et al., MicroRNA-582-5p suppresses non-small cell lung cancer cells growth and invasion via downregulating NOTCH1, PLoS One, 14 (2019), e0217652. |
| [29] | L. L. Wang, M. Zhang, miR-582-5p is a potential prognostic marker in human non-small cell lung cancer and functions as a tumor suppressor by targeting MAP3K2, Eur. Rev. Med. Pharmacol. Sci., 22 (2018), 7760-7767. |
| [30] | K. Skrzypek, M. Tertil, S. Golda, M. Ciesla, K. Weglarczyk, G. Collet, et al., Interplay between heme oxygenase-1 and miR-378 affects non-small cell lung carcinoma growth, vascularization, and metastasis, Antioxid. Redox Signaling, 19 (2013), 644-660. |
| [31] | H. Y. Lee, S. S. Han, H. Rhee, J. H. Park, J. S. Lee, Y. M. Oh, et al., Differential expression of microRNAs and their target genes in non-small-cell lung cancer, Mol. Med. Rep., 11 (2015), 2034-2040. |
| [32] | H. Y. Lee, S. S. Han, S. Y. Song, Serum microRNAs as potential biomarkers for lung cancer, Ann. Oncol., 27 (2016). |
| [33] | X. H. Wang, Y. Lu, J. J. Liang, J. X. Cao, Y. Q. Jin, G. S. An, et al., MiR-509-3-5p causes aberrant mitosis and anti-proliferative effect by suppression of PLK1 in human lung cancer A549 cells, Biochem. Biophys. Res. Commun., 478 (2016), 676-682. |
| [34] | F. Xing, S. Sharma, Y. Liu, Y. Y. Mo, K. Wu, Y. Y. Zhang, et al., miR-509 suppresses brain metastasis of breast cancer cells by modulating RhoC and TNF-α, Oncogene, 34 (2015), 4890. |
| [35] | Q. Zhai, L. Zhou, C. Zhao, J Wan, Z. Yu, X. Guo, et al., Identification of miR-508-3p and miR-509-3p that are associated with cell invasion and migration and involved in the apoptosis of renal cell carcinoma, Biochem. Biophys. Res. Commun., 419 (2012), 621-626. |
| [36] | G. Korkmaz, C. Le Sage, K. A. Tekirdag, R. Agami, D. Gozuacik, miR-376b controls starvation and mTOR inhibition-related autophagy by targeting ATG4C and BECN1, Autophagy, 8 (2012), 165-176. |
| [37] | D. Chen, W. Guo, Z. Qiu, Q. Wang, Y. Li, L. Liang, et al., MicroRNA-30d-5p inhibits tumour cell proliferation and motility by directly targeting CCNE2 in non-small cell lung cancer, Cancer Lett., 362 (2015), 208-217. |
| [38] | Y. Li, P. Chen, L. Zu, B. Liu, M. Wang, Q. Zhou, MicroRNA-338-3p suppresses metastasis of lung cancer cells by targeting the EMT regulator Sox4, Am. J. Cancer Res., 6 (2016), 127-140. |
| [39] | X. Chen, L. Wei, S. Zhao, miR-338 inhibits the metastasis of lung cancer by targeting integrin beta3, Oncol. Rep., 36 (2016), 1467-1474. |
| [40] | P. Zhang, G. Shao, X. Lin, Y. Liu, Z. Yang, MiR-338-3p inhibits the growth and invasion of non-small cell lung cancer cells by targeting IRS2, Am. J. Cancer Res., 7 (2017), 53-63. |
| [41] | J. Sui, S. Y. Xu, J. Han, S. R. Yang, C. Y. Li, L. H. Yin, et al., Integrated analysis of competing endogenous RNA network revealing lncRNAs as potential prognostic biomarkers in human lung squamous cell carcinoma, Oncotarget, 8 (2017), 65997-66018. |
| [42] | X. Zhu, S. Ju, F. Yuan, G. Chen, Y. Shu, C. Li, et al., microRNA-664 enhances proliferation, migration and invasion of lung cancer cells, Exp. The.r Med., 13 (2017), 3555-3562. |
| [43] | F. Yang, K. Wei, Z. Qin, W. Liu, C. Shao, C. Wang, et al., MiR-598 suppresses invasion and migration by negative regulation of derlin-1 and epithelial-mesenchymal transition in non-small cell lung cancer, Cell Physiol. Biochem., 47 (2018), 245-256. |
| [44] | X. Tong, P. Su, H. Yang, F. Chi, L. Shen, X. Feng, et al., MicroRNA-598 inhibits the proliferation and invasion of non-small cell lung cancer cells by directly targeting ZEB2, Exp. Ther. Med., 16 (2018), 5417-5423. |
| [45] | L. Xu, B. Wei, H. Hui, Y. Sun, Y. Liu, X. Yu, et al., Positive feedback loop of lncRNA LINC01296/miR-598/Twist1 promotes non-small cell lung cancer tumorigenesis, J. Cell Physiol., 234 (2019), 4563-4571. |
| [46] | J. Kim, N. J. Lim, S. G. Jang, H. K. Kim, G. K. Lee, miR-592 and miR-552 can distinguish between primary lung adenocarcinoma and colorectal cancer metastases in the lung, Anticancer Res., 34 (2014), 2297-2302. |
| [47] | J. Cao, X. R. Yan, T. Liu, X. B. Han, J. J. Yu, S. H. Liu, et al., MicroRNA-552 promotes tumor cell proliferation and migration by directly targeting DACH1 via the Wnt/beta-catenin signaling pathway in colorectal cancer, Oncol. Lett., 14 (2017), 3795-3802. |
| [48] | N. Wang, W. Liu, Increased expression of miR-552 acts as a potential predictor biomarker for poor prognosis of colorectal cancer, Eur. Rev. Med. Pharmacol. Sci., 22 (2018), 412-416. |
| [49] | M. Xu, Y. Z. Wang, miR133a suppresses cell proliferation, migration and invasion in human lung cancer by targeting MMP14, Oncol. Rep., 30 (2013), 1398-1404. |
| [50] | L. K. Wang, T. H. Hsiao, T. M. Hong, H. Y. Chen, S. H. Kao, W. L. Wang, et al., MicroRNA-133a suppresses multiple oncogenic membrane receptors and cell invasion in non-small cell lung carcinoma, PLoS One, 9 (2014), e96765. |
| [51] | Y. Wang, J. Li, H. Chen, Y. Mo, H. Ye, Y. Luo, et al., Down-regulation of miR-133a as a poor prognosticator in non-small cell lung cancer, Gene, 591 (2016), 333-337. |
| [52] | D. Schmitt, L. M. Da Silva, W. Zhang, Z. Liu, R. Arora, S. Lim, et al., ErbB2-intronic microRNA-4728: A novel tumor suppressor and antagonist of oncogenic MAPK signaling, Cell Death Dis., 6 (2015), e1742. |
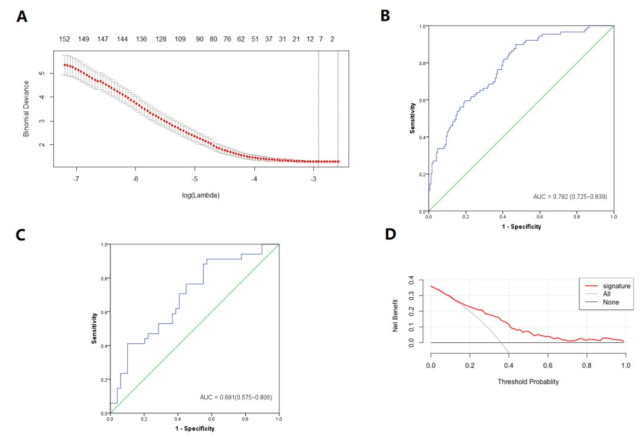
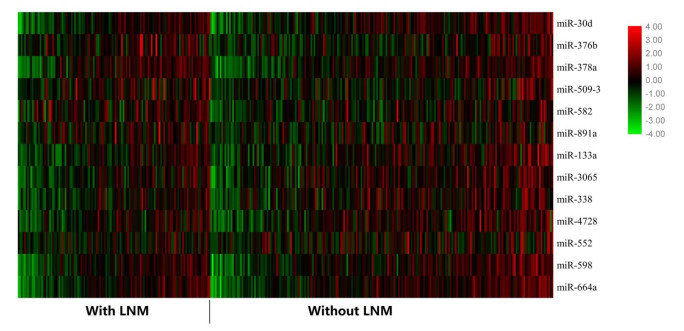
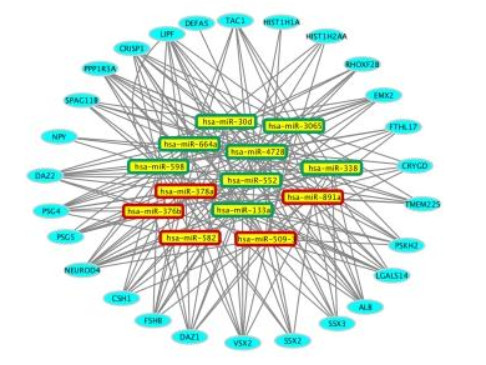
Figures(3) / Tables(2)

Shuyi Cen, Kaiyou Fu, Yue Shi, Hanliang Jiang, Jiawei Shou, Liangkun You, Weidong Han, Hongming Pan, Zhen Liu. A microRNA disease signature associated with lymph node metastasis of lung adenocarcinoma[J]. Mathematical Biosciences and Engineering, 2020, 17(3): 2557-2568. doi: 10.3934/mbe.2020140







 DownLoad:
DownLoad: