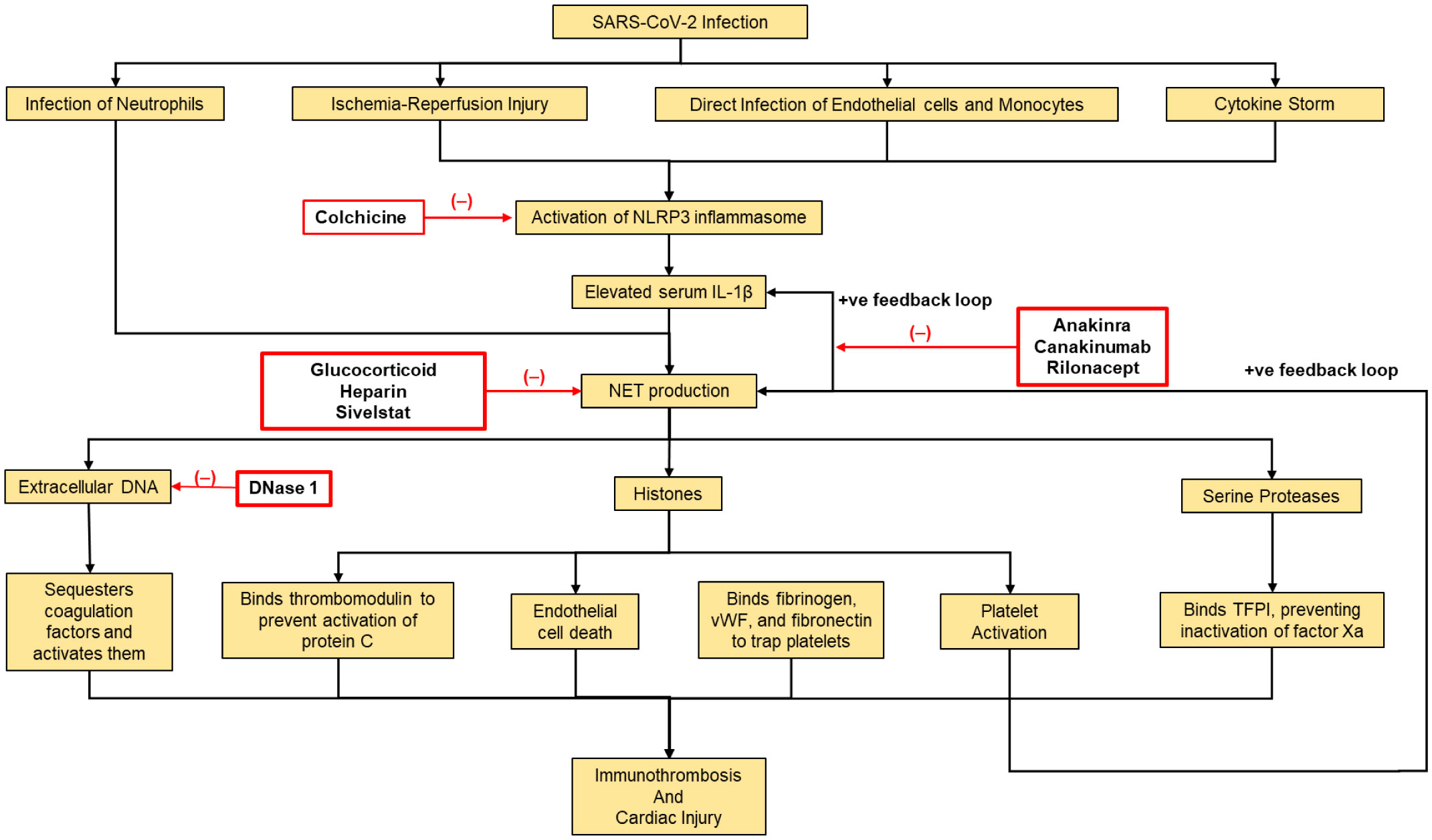
The COVID-19 pandemic has driven an upheaval of new research, providing key insights into the pathogenesis of this disease. Lymphocytopenia, hyper-inflammation and cardiac involvement are prominent features of the disease and have prognostic value. However, the mechanistic links among these phenomena are not well understood. Likewise, some COVID-19 patients exhibit multi-organ failure with diseases affecting the cardiac system, appearing to be an emerging feature of the COVID-19 pandemic. Neutrophil extracellular traps (NETs) have been frequently correlated with larger infarct sizes and can predict major adverse cardiac events. However, the exact mechanism behind this remains unknown. Although the excessive NET formation can drive inflammation, particularly endothelial and promote thrombosis, it is essential to normal immunity. In this paper, we postulate the role of NETs in cardiac disease by providing an overview of the relationship between NET and inflammasome activities in lung and liver diseases, speculating a link between these entities in cardiac diseases as well. Future research is required to specify the role of NETs in COVID-19, since this carries potential therapeutic significance, as inhibition of NETosis could alleviate symptoms of this disease. Knowledge gained from this could serve to inform the assessment and therapeutics of other hyper inflammatory diseases affecting the heart and vasculature alike.
Citation: Amal Feiroze Farouk, Areez Shafqat, Shameel Shafqat, Junaid Kashir, Khaled Alkattan, Ahmed Yaqinuddin. COVID-19 associated cardiac disease: Is there a role of neutrophil extracellular traps in pathogenesis?[J]. AIMS Molecular Science, 2021, 8(4): 275-290. doi: 10.3934/molsci.2021021
The COVID-19 pandemic has driven an upheaval of new research, providing key insights into the pathogenesis of this disease. Lymphocytopenia, hyper-inflammation and cardiac involvement are prominent features of the disease and have prognostic value. However, the mechanistic links among these phenomena are not well understood. Likewise, some COVID-19 patients exhibit multi-organ failure with diseases affecting the cardiac system, appearing to be an emerging feature of the COVID-19 pandemic. Neutrophil extracellular traps (NETs) have been frequently correlated with larger infarct sizes and can predict major adverse cardiac events. However, the exact mechanism behind this remains unknown. Although the excessive NET formation can drive inflammation, particularly endothelial and promote thrombosis, it is essential to normal immunity. In this paper, we postulate the role of NETs in cardiac disease by providing an overview of the relationship between NET and inflammasome activities in lung and liver diseases, speculating a link between these entities in cardiac diseases as well. Future research is required to specify the role of NETs in COVID-19, since this carries potential therapeutic significance, as inhibition of NETosis could alleviate symptoms of this disease. Knowledge gained from this could serve to inform the assessment and therapeutics of other hyper inflammatory diseases affecting the heart and vasculature alike.

| [1] |
Gupta A, Madhavan MV, Sehgal K, et al. (2020) Extrapulmonary manifestations of COVID-19. Nat Med 26: 1017-1032. doi: 10.1038/s41591-020-0968-3

|
| [2] |
Ramos-Casals M, Brito-Zerón P, Mariette X (2021) Systemic and organ-specific immune-related manifestations of COVID-19. Nat Rev Rheumatol 17: 315-332. doi: 10.1038/s41584-021-00608-z
|
| [3] |
Akhmerov A, Marbán E (2020) COVID-19 and the Heart. Circ Res 126: 1443-1455. doi: 10.1161/CIRCRESAHA.120.317055
|
| [4] |
Guo T, Fan Y, Chen M, et al. (2020) Cardiovascular Implications of Fatal Outcomes of Patients With Coronavirus Disease 2019 (COVID-19). JAMA Cardiol 5: 811-818. doi: 10.1001/jamacardio.2020.1017
|
| [5] |
Hilscher MB, Shah VH (2020) Neutrophil Extracellular Traps and Liver Disease. Semin Liver Dis 40: 171-179. doi: 10.1055/s-0039-3399562
|
| [6] |
El Kazzi M, Rayner BS, Chami B, et al. (2020) Neutrophil-Mediated Cardiac Damage After Acute Myocardial Infarction: Significance of Defining a New Target Cell Type for Developing Cardioprotective Drugs. Antioxid Redox Signal 33: 689-712. doi: 10.1089/ars.2019.7928
|
| [7] |
Yaqinuddin A, Kashir J (2020) Innate immunity in COVID-19 patients mediated by NKG2A receptors, and potential treatment using Monalizumab, Cholroquine, and antiviral agents. Med Hypotheses 140: 109777. doi: 10.1016/j.mehy.2020.109777
|
| [8] |
Yaqinuddin A, Kashir J (2020) Novel therapeutic targets for SARS-CoV-2-induced acute lung injury: Targeting a potential IL-1β/neutrophil extracellular traps feedback loop. Med Hypotheses 143: 109906. doi: 10.1016/j.mehy.2020.109906
|
| [9] |
Yaqinuddin A, Kvietys P, Kashir J (2020) COVID-19: Role of neutrophil extracellular traps in acute lung injury. Respir Investig 58: 419-420. doi: 10.1016/j.resinv.2020.06.001
|
| [10] |
Lee C, Choi WJ (2021) Overview of COVID-19 inflammatory pathogenesis from the therapeutic perspective. Arch Pharm Res 44: 99-116. doi: 10.1007/s12272-020-01301-7
|
| [11] |
Liao M, Liu Y, Yuan J, et al. (2020) Single-cell landscape of bronchoalveolar immune cells in patients with COVID-19. Nat Med 26: 842-844. doi: 10.1038/s41591-020-0901-9
|
| [12] |
Kvietys PR, Fakhoury HMA, Kadan S, et al. (2021) COVID-19: Lung-Centric Immunothrombosis. Front Cell Infect Microbiol 11: 679878. doi: 10.3389/fcimb.2021.679878
|
| [13] | Yaqinuddin A, Kashir J (2020) The central role of neutrophil extracellular traps in SARS-CoV-2-induced thrombogenesis and vasculitis. J Afr J Respir Med 15: 5. |
| [14] |
Ackermann M, Verleden SE, Kuehnel M, et al. (2020) Pulmonary Vascular Endothelialitis, Thrombosis, and Angiogenesis in Covid-19. N Engl J Med 383: 120-128. doi: 10.1056/NEJMoa2015432
|
| [15] |
Vasquez-Bonilla WO, Orozco R, Argueta V, et al. (2020) A review of the main histopathological findings in coronavirus disease 2019. Hum Pathol 105: 74-83. doi: 10.1016/j.humpath.2020.07.023
|
| [16] |
Rapkiewicz AV, Mai X, Carsons SE, et al. (2020) Megakaryocytes and platelet-fibrin thrombi characterize multi-organ thrombosis at autopsy in COVID-19: A case series. EClinicalMedicine 24: 100434. doi: 10.1016/j.eclinm.2020.100434
|
| [17] |
Agrati C, Sacchi A, Bordoni V, et al. (2020) Expansion of myeloid-derived suppressor cells in patients with severe coronavirus disease (COVID-19). Cell Death Differ 27: 3196-3207. doi: 10.1038/s41418-020-0572-6
|
| [18] |
Carissimo G, Xu W, Kwok I, et al. (2020) Whole blood immunophenotyping uncovers immature neutrophil-to-VD2 T-cell ratio as an early marker for severe COVID-19. Nat Commun 11: 5243. doi: 10.1038/s41467-020-19080-6
|
| [19] |
Mohamed Khosroshahi L, Rezaei N (2021) Dysregulation of the immune response in coronavirus disease 2019. Cell Biol Int 45: 702-707. doi: 10.1002/cbin.11517
|
| [20] | Luo XH, Zhu Y, Mao J, et al. (2021) T cell immunobiology and cytokine storm of COVID-19. Scand J Immunol 93: e12989. |
| [21] |
Wilk AJ, Rustagi A, Zhao NQ, et al. (2020) A single-cell atlas of the peripheral immune response in patients with severe COVID-19. Nat Med 26: 1070-1076. doi: 10.1038/s41591-020-0944-y
|
| [22] |
Ackermann M, Anders HJ, Bilyy R, et al. (2021) Patients with COVID-19: in the dark-NETs of neutrophils. Cell Death Differ 28: 3125-3139. doi: 10.1038/s41418-021-00805-z
|
| [23] |
Gollomp K, Kim M, Johnston I, et al. (2018) Neutrophil accumulation and NET release contribute to thrombosis in HIT. JCI Insight 3: e99445. doi: 10.1172/jci.insight.99445
|
| [24] |
Arcanjo A, Logullo J, Menezes CCB, et al. (2020) The emerging role of neutrophil extracellular traps in severe acute respiratory syndrome coronavirus 2 (COVID-19). Sci Rep 10: 19630. doi: 10.1038/s41598-020-76781-0
|
| [25] |
Ouwendijk WJD, Raadsen MP, van Kampen JJA, et al. (2021) High Levels of Neutrophil Extracellular Traps Persist in the Lower Respiratory Tract of Critically Ill Patients With Coronavirus Disease 2019. J Infect Dis 223: 1512-1521. doi: 10.1093/infdis/jiab050
|
| [26] |
Teluguakula N (2021) Neutrophils Set Extracellular Traps to Injure Lungs in Coronavirus Disease 2019. J Infect Dis 223: 1503-1505. doi: 10.1093/infdis/jiab053
|
| [27] |
Middleton EA, He XY, Denorme F, et al. (2020) Neutrophil extracellular traps contribute to immunothrombosis in COVID-19 acute respiratory distress syndrome. Blood 136: 1169-1179. doi: 10.1182/blood.2020007008
|
| [28] |
Schurink B, Roos E, Radonic T, et al. (2020) Viral presence and immunopathology in patients with lethal COVID-19: a prospective autopsy cohort study. Lancet Microbe 1: e290-e299. doi: 10.1016/S2666-5247(20)30144-0
|
| [29] |
Veras FP, Pontelli MC, Silva CM, et al. (2020) SARS-CoV-2-triggered neutrophil extracellular traps mediate COVID-19 pathology. J Exp Med 217: e20201129. doi: 10.1084/jem.20201129
|
| [30] |
Youn YJ, Lee YB, Kim SH, et al. (2021) Nucleocapsid and Spike Proteins of SARS-CoV-2 Drive Neutrophil Extracellular Trap Formation. Immune Netw 21: e16. doi: 10.4110/in.2021.21.e16
|
| [31] |
Iliadi V, Konstantinidou I, Aftzoglou K, et al. (2021) The Emerging Role of Neutrophils in the Pathogenesis of Thrombosis in COVID-19. Int J Mol Sci 22: 5368. doi: 10.3390/ijms22105368
|
| [32] |
Janiuk K, Jabłońska E, Garley M (2021) Significance of NETs Formation in COVID-19. Cells 10: 151. doi: 10.3390/cells10010151
|
| [33] |
Leshner M, Wang S, Lewis C, et al. (2012) PAD4 mediated histone hypercitrullination induces heterochromatin decondensation and chromatin unfolding to form neutrophil extracellular trap-like structures. Front Immunol 3: 307. doi: 10.3389/fimmu.2012.00307
|
| [34] |
Yipp BG, Petri B, Salina D, et al. (2012) Infection-induced NETosis is a dynamic process involving neutrophil multitasking in vivo. Nat Med 18: 1386-1393. doi: 10.1038/nm.2847
|
| [35] |
Leppkes M, Maueröder C, Hirth S, et al. (2016) Externalized decondensed neutrophil chromatin occludes pancreatic ducts and drives pancreatitis. Nat Commun 7: 10973. doi: 10.1038/ncomms10973
|
| [36] |
Giustino G, Croft LB, Stefanini GG, et al. (2020) Characterization of Myocardial Injury in Patients With COVID-19. J Am Coll Cardiol 76: 2043-2055. doi: 10.1016/j.jacc.2020.08.069
|
| [37] |
Fuchs TA, Brill A, Duerschmied D, et al. (2010) Extracellular DNA traps promote thrombosis. Proc Natl Acad Sci U S A 107: 15880-15885. doi: 10.1073/pnas.1005743107
|
| [38] |
Semeraro F, Ammollo CT, Morrissey JH, et al. (2011) Extracellular histones promote thrombin generation through platelet-dependent mechanisms: involvement of platelet TLR2 and TLR4. Blood 118: 1952-1961. doi: 10.1182/blood-2011-03-343061
|
| [39] |
Martinod K, Wagner DD (2014) Thrombosis: tangled up in NETs. Blood 123: 2768-2776. doi: 10.1182/blood-2013-10-463646
|
| [40] |
Fuchs TA, Bhandari AA, Wagner DD (2011) Histones induce rapid and profound thrombocytopenia in mice. Blood 118: 3708-3714. doi: 10.1182/blood-2011-01-332676
|
| [41] |
Xu J, Zhang X, Pelayo R, et al. (2009) Extracellular histones are major mediators of death in sepsis. Nat Med 15: 1318-1321. doi: 10.1038/nm.2053
|
| [42] |
Etulain J, Martinod K, Wong SL, et al. (2015) P-selectin promotes neutrophil extracellular trap formation in mice. Blood 126: 242-246. doi: 10.1182/blood-2015-01-624023
|
| [43] |
Maugeri N, Campana L, Gavina M, et al. (2014) Activated platelets present high mobility group box 1 to neutrophils, inducing autophagy and promoting the extrusion of neutrophil extracellular traps. J Thromb Haemost 12: 2074-2088. doi: 10.1111/jth.12710
|
| [44] |
Thålin C, Hisada Y, Lundström S, et al. (2019) Neutrophil Extracellular Traps: Villains and Targets in Arterial, Venous, and Cancer-Associated Thrombosis. Arterioscler Thromb Vasc Biol 39: 1724-1738. doi: 10.1161/ATVBAHA.119.312463
|
| [45] |
Boeltz S, Amini P, Anders HJ, et al. (2019) To NET or not to NET:current opinions and state of the science regarding the formation of neutrophil extracellular traps. Cell Death Differ 26: 395-408. doi: 10.1038/s41418-018-0261-x
|
| [46] |
Klopf J, Brostjan C, Eilenberg W, et al. (2021) Neutrophil Extracellular Traps and Their Implications in Cardiovascular and Inflammatory Disease. Int J Mol Sci 22: 559. doi: 10.3390/ijms22020559
|
| [47] |
Massberg S, Grahl L, von Bruehl ML, et al. (2010) Reciprocal coupling of coagulation and innate immunity via neutrophil serine proteases. Nat Med 16: 887-896. doi: 10.1038/nm.2184
|
| [48] |
Ammollo CT, Semeraro F, Xu J, et al. (2011) Extracellular histones increase plasma thrombin generation by impairing thrombomodulin-dependent protein C activation. J Thromb Haemost 9: 1795-1803. doi: 10.1111/j.1538-7836.2011.04422.x
|
| [49] |
Kang Y, Chen T, Mui D, et al. (2020) Cardiovascular manifestations and treatment considerations in COVID-19. Heart 106: 1132-1141. doi: 10.1136/heartjnl-2020-317056
|
| [50] |
Bearse M, Hung YP, Krauson AJ, et al. (2021) Factors associated with myocardial SARS-CoV-2 infection, myocarditis, and cardiac inflammation in patients with COVID-19. Mod Pathol 34: 1345-1357. doi: 10.1038/s41379-021-00790-1
|
| [51] |
Lindner D, Fitzek A, Bräuninger H, et al. (2020) Association of Cardiac Infection With SARS-CoV-2 in Confirmed COVID-19 Autopsy Cases. JAMA Cardiol 5: 1281-1285. doi: 10.1001/jamacardio.2020.3551
|
| [52] |
Van Linthout S, Miteva K, Tschöpe C (2014) Crosstalk between fibroblasts and inflammatory cells. Cardiovasc Res 102: 258-269. doi: 10.1093/cvr/cvu062
|
| [53] |
Lippi G, Plebani M (2020) Laboratory abnormalities in patients with COVID-2019 infection. Clin Chem Lab Med 58: 1131-1134. doi: 10.1515/cclm-2020-0198
|
| [54] | Blasco A, Coronado MJ, Hernández-Terciado F, et al. (2020) Assessment of Neutrophil Extracellular Traps in Coronary Thrombus of a Case Series of Patients With COVID-19 and Myocardial Infarction. JAMA Cardiol 6: 1-6. |
| [55] |
Henry BM, Aggarwal G, Wong J, et al. (2020) Lactate dehydrogenase levels predict coronavirus disease 2019 (COVID-19) severity and mortality: A pooled analysis. Am J Emerg Med 38: 1722-1726. doi: 10.1016/j.ajem.2020.05.073
|
| [56] |
Henry BM, de Oliveira MHS, Benoit S, et al. (2020) Hematologic, biochemical and immune biomarker abnormalities associated with severe illness and mortality in coronavirus disease 2019 (COVID-19): a meta-analysis. Clin Chem Lab Med 58: 1021-1028. doi: 10.1515/cclm-2020-0369
|
| [57] |
Szarpak L, Ruetzler K, Safiejko K, et al. (2021) Lactate dehydrogenase level as a COVID-19 severity marker. Am J Emerg Med 45: 638-639. doi: 10.1016/j.ajem.2020.11.025
|
| [58] |
Bonow RO, Fonarow GC, O'Gara PT, et al. (2020) Association of Coronavirus Disease 2019 (COVID-19) With Myocardial Injury and Mortality. JAMA Cardiol 5: 751-753. doi: 10.1001/jamacardio.2020.1105
|
| [59] |
Muraro SP, De Souza GF, Gallo SW, et al. (2018) Respiratory Syncytial Virus induces the classical ROS-dependent NETosis through PAD-4 and necroptosis pathways activation. Sci Rep 8: 14166. doi: 10.1038/s41598-018-32576-y
|
| [60] |
López-Reyes A, Martinez-Armenta C, Espinosa-Velázquez R, et al. (2020) NLRP3 Inflammasome: The Stormy Link Between Obesity and COVID-19. Front Immunol 11: 570251. doi: 10.3389/fimmu.2020.570251
|
| [61] |
Yang Y, Wang H, Kouadir M, et al. (2019) Recent advances in the mechanisms of NLRP3 inflammasome activation and its inhibitors. Cell Death Dis 10: 128. doi: 10.1038/s41419-019-1413-8
|
| [62] |
Lara PC, Macías-Verde D, Burgos-Burgos J (2020) Age-induced NLRP3 Inflammasome Over-activation Increases Lethality of SARS-CoV-2 Pneumonia in Elderly Patients. Aging Dis 11: 756-762. doi: 10.14336/AD.2020.0601
|
| [63] |
Gedefaw L, Ullah S, Leung PHM, et al. (2021) Inflammasome Activation-Induced Hypercoagulopathy: Impact on Cardiovascular Dysfunction Triggered in COVID-19 Patients. Cells 10: 916. doi: 10.3390/cells10040916
|
| [64] |
Saeedi-Boroujeni A, Mahmoudian-Sani MR, Nashibi R, et al. (2021) Tranilast: a potential anti-Inflammatory and NLRP3 inflammasome inhibitor drug for COVID-19. Immunopharmacol Immunotoxicol 43: 247-258. doi: 10.1080/08923973.2021.1925293
|
| [65] |
Ng H, Havervall S, Rosell A, et al. (2021) Circulating Markers of Neutrophil Extracellular Traps Are of Prognostic Value in Patients With COVID-19. Arterioscler Thromb Vasc Biol 41: 988-994. doi: 10.1161/ATVBAHA.120.315267
|
| [66] |
Bonaventura A, Vecchié A, Abbate A, et al. (2020) Neutrophil Extracellular Traps and Cardiovascular Diseases: An Update. Cells 9: 231. doi: 10.3390/cells9010231
|
| [67] |
Distelmaier K, Winter MP, Dragschitz F, et al. (2014) Prognostic value of culprit site neutrophils in acute coronary syndrome. Eur J Clin Invest 44: 257-265. doi: 10.1111/eci.12228
|
| [68] |
Ge L, Zhou X, Ji WJ, et al. (2015) Neutrophil extracellular traps in ischemia-reperfusion injury-induced myocardial no-reflow: therapeutic potential of DNase-based reperfusion strategy. Am J Physiol Heart Circ Physiol 308: H500-509. doi: 10.1152/ajpheart.00381.2014
|
| [69] |
Huang H, Tohme S, Al-Khafaji AB, et al. (2015) Damage-associated molecular pattern-activated neutrophil extracellular trap exacerbates sterile inflammatory liver injury. Hepatology 62: 600-614. doi: 10.1002/hep.27841
|
| [70] |
Kawaguchi M, Takahashi M, Hata T, et al. (2011) Inflammasome activation of cardiac fibroblasts is essential for myocardial ischemia/reperfusion injury. Circulation 123: 594-604. doi: 10.1161/CIRCULATIONAHA.110.982777
|
| [71] |
Giannopoulos G, Vrachatis DA, Deftereos SG (2020) Myocardial Injury in COVID-19-Can We Successfully Target Inflammation? JAMA Cardiol 5: 1069-1070. doi: 10.1001/jamacardio.2020.2569
|
| [72] |
Liu J, Li J, Arnold K, et al. (2020) Using heparin molecules to manage COVID-2019. Res Pract Thromb Haemost 4: 518-523. doi: 10.1002/rth2.12353
|
| [73] |
Horby P, Lim WS, Emberson JR, et al. (2021) Dexamethasone in Hospitalized Patients with Covid-19. N Engl J Med 384: 693-704. doi: 10.1056/NEJMoa2021436
|
| [74] |
Boulware DR, Pullen MF, Bangdiwala AS, et al. (2020) A Randomized Trial of Hydroxychloroquine as Postexposure Prophylaxis for Covid-19. N Engl J Med 383: 517-525. doi: 10.1056/NEJMoa2016638
|
| [75] |
Sun Y, Chen C, Zhang X, et al. (2020) Heparin improves alveolarization and vascular development in hyperoxia-induced bronchopulmonary dysplasia by inhibiting neutrophil extracellular traps. Biochem Biophys Res Commun 522: 33-39. doi: 10.1016/j.bbrc.2019.11.041
|
| [76] |
Santocki M, Kolaczkowska E (2020) On Neutrophil Extracellular Trap (NET) Removal: What We Know Thus Far and Why So Little. Cells 9: 2079. doi: 10.3390/cells9092079
|
| [77] | Kashir J, Ambia AR, Shafqat A, et al. (2021) Scientific premise for the involvement of neutrophil extracellular traps (NETs) in vaccine-induced thrombotic thrombocytopenia (VITT). J Leukoc Biol . |
| [78] |
Weber AG, Chau AS, Egeblad M, et al. (2020) Nebulized in-line endotracheal dornase alfa and albuterol administered to mechanically ventilated COVID-19 patients: a case series. Mol Med 26: 91. doi: 10.1186/s10020-020-00215-w
|
| [79] |
Laridan E, Denorme F, Desender L, et al. (2017) Neutrophil extracellular traps in ischemic stroke thrombi. Ann Neurol 82: 223-232. doi: 10.1002/ana.24993
|
| [80] |
Bikdeli B, Madhavan MV, Gupta A, et al. (2020) Pharmacological Agents Targeting Thromboinflammation in COVID-19: Review and Implications for Future Research. Thromb Haemost 120: 1004-1024. doi: 10.1055/s-0040-1713152
|
| [81] |
Simka M (2021) Is digital necrosis in COVID-19 caused by neutrophil extracellular traps: Potential therapeutic strategies. Med Hypotheses 156: 110684. doi: 10.1016/j.mehy.2021.110684
|
| [82] |
Gillot C, Favresse J, Mullier F, et al. (2021) NETosis and the Immune System in COVID-19: Mechanisms and Potential Treatments. Front Pharmacol 12: 708302. doi: 10.3389/fphar.2021.708302
|
| [83] |
Okubo K, Kamiya M, Urano Y, et al. (2016) Lactoferrin Suppresses Neutrophil Extracellular Traps Release in Inflammation. EBioMedicine 10: 204-215. doi: 10.1016/j.ebiom.2016.07.012
|
| [84] |
Liu X, Li Z, Liu S, et al. (2020) Potential therapeutic effects of dipyridamole in the severely ill patients with COVID-19. Acta Pharm Sin B 10: 1205-1215. doi: 10.1016/j.apsb.2020.04.008
|
| [85] |
Ali RA, Gandhi AA, Meng H, et al. (2019) Adenosine receptor agonism protects against NETosis and thrombosis in antiphospholipid syndrome. Nat Commun 10: 1916. doi: 10.1038/s41467-019-09801-x
|
| [86] |
Hazeldine J, Lord JM (2021) Neutrophils and COVID-19: Active Participants and Rational Therapeutic Targets. Front Immunol 12: 680134. doi: 10.3389/fimmu.2021.680134
|
| [87] |
Kyriazopoulou E, Poulakou G, Milionis H, et al. (2021) Early treatment of COVID-19 with anakinra guided by soluble urokinase plasminogen receptor plasma levels: a double-blind, randomized controlled phase 3 trial. Nat Med 27: 1752-1760. doi: 10.1038/s41591-021-01499-z
|
| [88] |
Barnes BJ, Adrover JM, Baxter-Stoltzfus A, et al. (2020) Targeting potential drivers of COVID-19: Neutrophil extracellular traps. J Exp Med 217: e20200652. doi: 10.1084/jem.20200652
|
| [89] |
Pal D, Goyal J, Sharma U, et al. (2021) Mesenchymal stem cells in SARS-CoV-2 infection: A hype or hope. Life Sci 284: 119901. doi: 10.1016/j.lfs.2021.119901
|
| [90] |
Klok FA, Kruip M, van der Meer NJM, et al. (2020) Incidence of thrombotic complications in critically ill ICU patients with COVID-19. Thromb Res 191: 145-147. doi: 10.1016/j.thromres.2020.04.013
|
Figures(1)

Amal Feiroze Farouk, Areez Shafqat, Shameel Shafqat, Junaid Kashir, Khaled Alkattan, Ahmed Yaqinuddin. COVID-19 associated cardiac disease: Is there a role of neutrophil extracellular traps in pathogenesis?[J]. AIMS Molecular Science, 2021, 8(4): 275-290. doi: 10.3934/molsci.2021021







 DownLoad:
DownLoad: