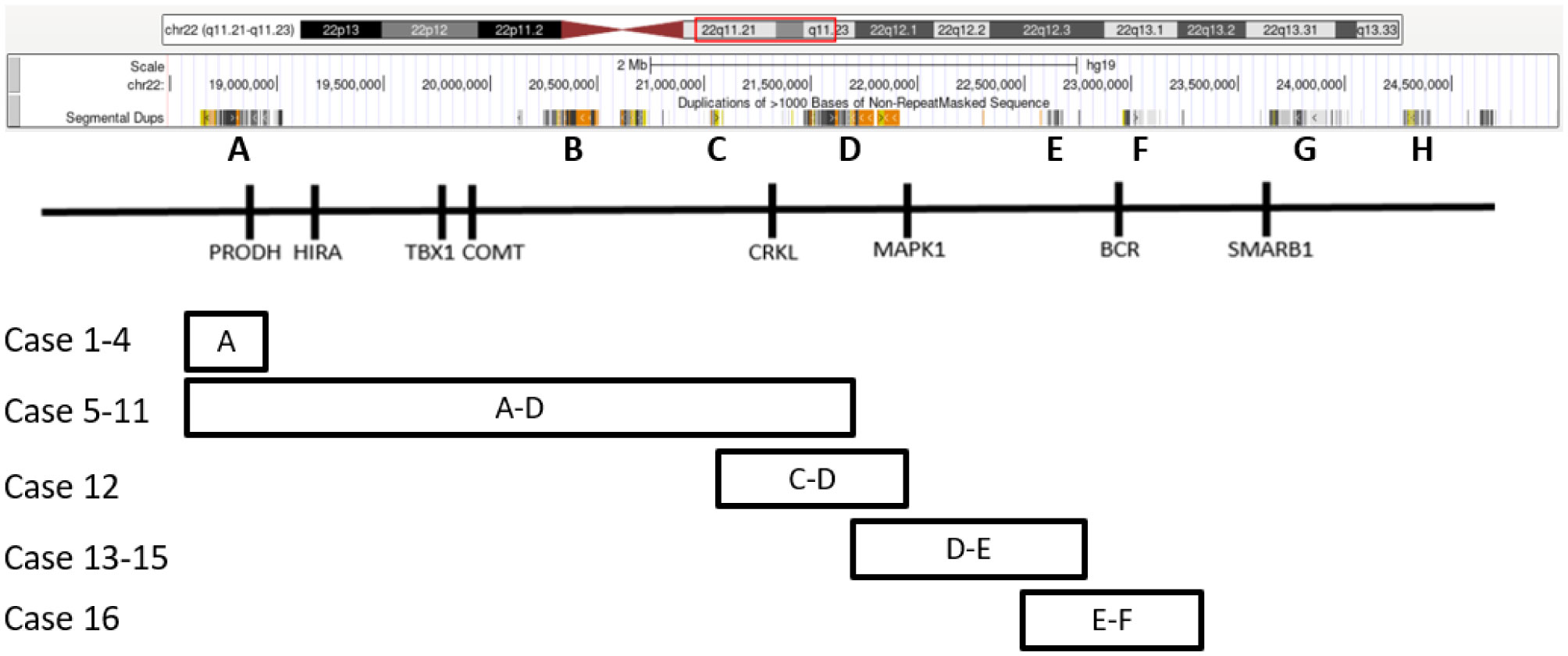
Patients with chromosome 22q11.2 deletion syndromes classically present with variable cardiac defects, parathyroid and thyroid gland hypoplasia, immunodeficiency and velopharyngeal insufficiency, developmental delay, intellectual disability, cognitive impairment, and psychiatric disorders. New technologies including chromosome microarray have identified smaller deletions in the 22q11.2 region. An increasing number of studies have reported patients presenting with various features harboring smaller 22q11.2 deletions, suggesting a need to better elucidate 22q11.2 deletions and their phenotypic contributions so that clinicians may better guide prognosis for families. We identified 16 pediatric patients at our institution harboring various 22q11.2 deletions detected by chromosomal microarray and report their clinical presentations. Findings include various neurodevelopmental delays with the most common one being attention deficit hyperactivity disorder (ADHD), one reported case of infant lethality, four cases of preterm birth, one case with dual diagnoses of 22q11.2 microdeletion and Down syndrome. We examined potential genotypic contributions of the deleted regions.
Citation: Gabrielle C. Manno, Gabrielle S. Segal, Alexander Yu, Fangling Xu, Joseph W. Ray, Erin Cooney, Allison D. Britt, Sunil K. Jain, Randall M. Goldblum, Sally S. Robinson, Jianli Dong. Genotypic and phenotypic variability of 22q11.2 microdeletions – an institutional experience[J]. AIMS Molecular Science, 2021, 8(4): 257-274. doi: 10.3934/molsci.2021020
Patients with chromosome 22q11.2 deletion syndromes classically present with variable cardiac defects, parathyroid and thyroid gland hypoplasia, immunodeficiency and velopharyngeal insufficiency, developmental delay, intellectual disability, cognitive impairment, and psychiatric disorders. New technologies including chromosome microarray have identified smaller deletions in the 22q11.2 region. An increasing number of studies have reported patients presenting with various features harboring smaller 22q11.2 deletions, suggesting a need to better elucidate 22q11.2 deletions and their phenotypic contributions so that clinicians may better guide prognosis for families. We identified 16 pediatric patients at our institution harboring various 22q11.2 deletions detected by chromosomal microarray and report their clinical presentations. Findings include various neurodevelopmental delays with the most common one being attention deficit hyperactivity disorder (ADHD), one reported case of infant lethality, four cases of preterm birth, one case with dual diagnoses of 22q11.2 microdeletion and Down syndrome. We examined potential genotypic contributions of the deleted regions.

| [1] |
Burnside RD (2015) 22q11.21 Deletion Syndromes: A Review of Proximal, Central, and Distal Deletions and Their Associated Features. Cytogenet Genome Res 146: 89-99. doi: 10.1159/000438708

|
| [2] |
Gerdes M, Solot C, Wang PP, et al. (1999) Cognitive and behavior profile of preschool children with chromosome 22q11.2 deletion. Am J Med Genet 85: 127-133. doi: 10.1002/(SICI)1096-8628(19990716)85:2<127::AID-AJMG6>3.0.CO;2-F
|
| [3] | McDonald-McGinn DM, Hain HS, Emanuel BS, et al. (1993) 22q11.2 Deletion Syndrome. GeneReviews((R)) Seattle (WA): University of Washington, Seattle. |
| [4] |
McDonald-McGinn DM, Sullivan KE, Marino B, et al. (2015) 22q11.2 deletion syndrome. Nat Rev Dis Primers 1: 15071. doi: 10.1038/nrdp.2015.71
|
| [5] |
Moss EM, Batshaw ML, Solot CB, et al. (1999) Psychoeducational profile of the 22q11.2 microdeletion: A complex pattern. J Pediatr 134: 193-198. doi: 10.1016/S0022-3476(99)70415-4
|
| [6] |
Shprintzen RJ, Goldberg R, Golding-Kushner KJ, et al. (1992) Late-onset psychosis in the velo-cardio-facial syndrome. Am J Med Genet 42: 141-142. doi: 10.1002/ajmg.1320420131
|
| [7] |
Leite AJ, Pinto IP, Cunha DM, et al. (2016) The Identification of Microdeletion and Reciprocal Microduplication in 22q11.2 Using High-Resolution CMA Technology. Biomed Res Int 2016: 7415438. doi: 10.1155/2016/7415438
|
| [8] |
Davies RW, Fiksinski AM, Breetvelt EJ, et al. (2020) Using common genetic variation to examine phenotypic expression and risk prediction in 22q11.2 deletion syndrome. Nat Med 26: 1912-1918. doi: 10.1038/s41591-020-1103-1
|
| [9] |
Homans JF, Tromp IN, Colo D, et al. (2018) Orthopaedic manifestations within the 22q11.2 Deletion syndrome: A systematic review. Am J Med Genet A 176: 2104-2120. doi: 10.1002/ajmg.a.38545
|
| [10] |
Rump P, de Leeuw N, van Essen AJ, et al. (2014) Central 22q11.2 deletions. Am J Med Genet A 164A: 2707-2723. doi: 10.1002/ajmg.a.36711
|
| [11] |
Verhagen JM, Diderich KE, Oudesluijs G, et al. (2012) Phenotypic variability of atypical 22q11.2 deletions not including TBX1. Am J Med Genet A 158A: 2412-2420. doi: 10.1002/ajmg.a.35517
|
| [12] |
Yu A, Turbiville D, Xu F, et al. (2019) Genotypic and phenotypic variability of 22q11.2 microduplications: An institutional experience. Am J Med Genet A 179: 2178-2189. doi: 10.1002/ajmg.a.61345
|
| [13] |
Riggs ER, Andersen EF, Cherry AM, et al. (2020) Technical standards for the interpretation and reporting of constitutional copy-number variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics (ACMG) and the Clinical Genome Resource (ClinGen). Genet Med 22: 245-257. doi: 10.1038/s41436-019-0686-8
|
| [14] |
Kearney HM, Thorland EC, Brown KK, et al. (2011) Working Group of the American College of Medical Genetics Laboratory Quality Assurance, C. American College of Medical Genetics standards and guidelines for interpretation and reporting of postnatal constitutional copy number variants. Genet Med 13: 680-685. doi: 10.1097/GIM.0b013e3182217a3a
|
| [15] |
Tarquinio DC, Jones MC, Jones KL, et al. (2012) Growth Charts for 22q11 Deletion Syndrome. Am J Med Genet Part A 58: 2672-2681. doi: 10.1002/ajmg.a.35485
|
| [16] | Abd El-Ghany HM, Mekkawy MK, Helmy NA, et al. (2018) Molecular characterization of patients with clinical suspicion of 22q11.2 deletion syndrome. Middle East J Med Genet 7: 32-38. |
| [17] |
Miller DT, Adam MP, Aradhya S, et al. (2010) Consensus statement: chromosomal microarray is a first-tier clinical diagnostic test for individuals with developmental disabilities or congenital anomalies. Am J Hum Genet 86: 749-764. doi: 10.1016/j.ajhg.2010.04.006
|
| [18] |
Crowley B, Ruffner M, Mcginn DM, et al. (2018) Variable immune deficiency related to deletion size in chromosome 22q11.2 deletion syndrome. Am J Med Genet A 176: 2082-2086. doi: 10.1002/ajmg.a.38597
|
| [19] |
Mikhail FM, Burnside RD, Rush B, et al. (2014) The recurrent distal 22q11.2 microdeletions are often de novo and do not represent a single clinical entity: a proposed categorization system. Genet Med 16: 92-100. doi: 10.1038/gim.2013.79
|
| [20] |
Motahari Z, Moody SA, Maynard TM, et al. (2019) In the line-up: deleted genes associated with DiGeorge/22q11.2 deletion syndrome: are they all suspects? J Neurodev Disord 11: 7. doi: 10.1186/s11689-019-9267-z
|
| [21] |
Zhao Y, Guo T, Fiksinski A, et al. (2018) Variance of IQ is partially dependent on deletion type among 1,427 22q11.2 deletion syndrome subjects. Am J Med Genet A 176: 2172-2181. doi: 10.1002/ajmg.a.40359
|
| [22] |
Cirillo E, Giardino G, Gallo V, et al. (2014) Intergenerational and intrafamilial phenotypic variability in 22q11.2 deletion syndrome subjects. BMC Med Genet 15: 1. doi: 10.1186/1471-2350-15-1
|
| [23] |
Ramachandran D, Mulle JG, Locke AE, et al. (2015) Contribution of copy-number variation to Down syndrome-associated atrioventricular septal defects. Genet Med 17: 554-560. doi: 10.1038/gim.2014.144
|
| [24] |
Guo T, Diacou A, Nomaru H, et al. (2018) Deletion size analysis of 1680 22q11.2DS subjects identifies a new recombination hotspot on chromosome 22q11.2. Hum Mol Genet 27: 1150-1163. doi: 10.1093/hmg/ddy028
|
| [25] |
Das Chakraborty R, Bernal AJ, Schoch K, et al. (2012) Dysregulation of DGCR6 and DGCR6L: psychopathological outcomes in chromosome 22q11.2 deletion syndrome. Transl Psychiatry 2: e105. doi: 10.1038/tp.2012.31
|
| [26] |
Du Q, de la Morena MT, van Oers NSC (2019) The Genetics and Epigenetics of 22q11.2 Deletion Syndrome. Front Genet 10: 1365. doi: 10.3389/fgene.2019.01365
|
| [27] |
Jacquet H, Raux G, Thibaut F, et al. (2002) PRODH mutations and hyperprolinemia in a subset of schizophrenic patients. Hum Mol Genet 11: 2243-2249. doi: 10.1093/hmg/11.19.2243
|
| [28] |
Kempf L, Nicodemus KK, Kolachana B, et al. (2008) Functional polymorphisms in PRODH are associated with risk and protection for schizophrenia and fronto-striatal structure and function. PLoS Genet 4: e1000252. doi: 10.1371/journal.pgen.1000252
|
| [29] |
Namavar Y, Duineveld DJ, Both GIA, et al. (2021) Psychiatric phenotypes associated with hyperprolinemia: A systematic review. Am J Med Genet B Neuropsychiatr Genet 186: 289-317. doi: 10.1002/ajmg.b.32869
|
| [30] |
Raux G, Bumsel E, Hecketsweiler B, et al. (2007) Involvement of hyperprolinemia in cognitive and psychiatric features of the 22q11 deletion syndrome. Hum Mol Genet 16: 83-91. doi: 10.1093/hmg/ddl443
|
| [31] |
Richard AC, Rovelet-Lecrux A, Delaby E, et al. (2016) The 22q11 PRODH/DGCR6 deletion is frequent in hyperprolinemic subjects but is not a strong risk factor for ASD. Am J Med Genet B Neuropsychiatr Genet 171B: 377-382. doi: 10.1002/ajmg.b.32416
|
| [32] |
Fiksinski AM, Schneider M, Murphy CM, et al. (2018) Understanding the pediatric psychiatric phenotype of 22q11.2 deletion syndrome. Am J Med Genet A 176: 2182-2191. doi: 10.1002/ajmg.a.40387
|
| [33] |
Niklasson L, Rasmussen P, Oskarsdottir S, et al. (2009) Autism, ADHD, mental retardation and behavior problems in 100 individuals with 22q11 deletion syndrome. Res Dev Disabil 30: 763-773. doi: 10.1016/j.ridd.2008.10.007
|
| [34] |
Serur Y, Sofrin Frumer D, Daon K, et al. (2019) Psychiatric disorders and autism in young children with 22q11.2 deletion syndrome compared to children with idiopathic autism. Eur Psychiatry 55: 116-121. doi: 10.1016/j.eurpsy.2018.10.007
|
| [35] |
Taylor LE, Kates WR, Fremont W, et al. (2018) Young Adult Outcomes for Children With 22q11 Deletion Syndrome and Comorbid ADHD. J Pediatr Psychol 43: 636-644. doi: 10.1093/jpepsy/jsy002
|
| [36] |
Schindewolf E, Khalek N, Johnson MP, et al. (2018) Expanding the fetal phenotype: Prenatal sonographic findings and perinatal outcomes in a cohort of patients with a confirmed 22q11.2 deletion syndrome. Am J Med Genet A 176: 1735-1741. doi: 10.1002/ajmg.a.38665
|
| [37] |
Beleza-Meireles A, Clayton-Smith J, Saraiva JM, et al. (2014) Oculo-auriculo-vertebral spectrum: a review of the literature and genetic update. J Med Genet 51: 635-645. doi: 10.1136/jmedgenet-2014-102476
|
| [38] |
Bogusiak K, Puch A, Arkuszewski P (2017) Goldenhar syndrome: current perspectives. World J Pediatr 13: 405-415. doi: 10.1007/s12519-017-0048-z
|
| [39] |
Spineli-Silva S, Bispo LM, Gil-da-Silva-Lopes VL, et al. (2018) Distal deletion at 22q11.2 as differential diagnosis in Craniofacial Microsomia: Case report and literature review. Eur J Med Genet 61: 262-268. doi: 10.1016/j.ejmg.2017.12.013
|
| [40] |
Spineli-Silva S, Sgardioli IC, Dos Santos AP, et al. (2020) Genomic imbalances in craniofacial microsomia. Am J Med Genet C Semin Med Genet 184: 970-985. doi: 10.1002/ajmg.c.31857
|
| [41] |
Rozas MF, Benavides F, Leon L, et al. (2019) Association between phenotype and deletion size in 22q11.2 microdeletion syndrome: systematic review and meta-analysis. Orphanet J Rare Dis 14: 195. doi: 10.1186/s13023-019-1170-x
|
| [42] |
Hiroi N, Takahashi T, Hishimoto A, et al. (2013) Copy number variation at 22q11.2: from rare variants to common mechanisms of developmental neuropsychiatric disorders. Mol Psychiatry 18: 1153-1165. doi: 10.1038/mp.2013.92
|
| [43] |
Jensen M, Kooy RF, Simon TJ, et al. (2018) A higher rare CNV burden in the genetic background potentially contributes to intellectual disability phenotypes in 22q11.2 deletion syndrome. Eur J Med Genet 61: 209-212. doi: 10.1016/j.ejmg.2017.11.016
|
| [44] |
Mlynarski EE, Xie M, Taylor D, et al. (2016) Rare copy number variants and congenital heart defects in the 22q11.2 deletion syndrome. Hum Genet 135: 273-285. doi: 10.1007/s00439-015-1623-9
|
Figures(1) / Tables(4)

Gabrielle C. Manno, Gabrielle S. Segal, Alexander Yu, Fangling Xu, Joseph W. Ray, Erin Cooney, Allison D. Britt, Sunil K. Jain, Randall M. Goldblum, Sally S. Robinson, Jianli Dong. Genotypic and phenotypic variability of 22q11.2 microdeletions – an institutional experience[J]. AIMS Molecular Science, 2021, 8(4): 257-274. doi: 10.3934/molsci.2021020







 DownLoad:
DownLoad: