Citation: Audrey Lejart, Gilles Salbert, Sébastien Huet. Cytosine hydroxymethylation by TET enzymes: From the control of gene expression to the regulation of DNA repair mechanisms, and back[J]. AIMS Biophysics, 2018, 5(3): 182-193. doi: 10.3934/biophy.2018.3.182

| [1] |
Geiman TM, Robertson KD (2002) Chromatin remodeling, histone modifications, and DNA methylation-how does it all fit together? J Cell Biochem 87: 117–125. doi: 10.1002/jcb.10286

|
| [2] |
Bonev B, Cavalli G (2016) Organization and function of the 3D genome. Nat Rev Genet 17: 661–678. doi: 10.1038/nrg.2016.112
|
| [3] |
Bannister AJ, Kouzarides T (2011) Regulation of chromatin by histone modifications. Cell Res 21: 381–395. doi: 10.1038/cr.2011.22
|
| [4] | Tessarz P, Kouzarides T (2014) Histone core modifications regulating nucleosome structure and dynamics. Nat Rev Mol Cell Bio 15: 703–708. |
| [5] |
Koch A, Joosten SC, Feng Z, et al. (2018) Analysis of DNA methylation in cancer: Location revisited. Nat Rev Clin Oncol 15: 459–466. doi: 10.1038/s41571-018-0004-4
|
| [6] |
Watt F, Molloy PL (1988) Cytosine methylation prevents binding to DNA of a HeLa cell transcription factor required for optimal expression of the adenovirus major late promoter. Gene Dev 2: 1136–1143. doi: 10.1101/gad.2.9.1136
|
| [7] |
Csankovszki G, Nagy A, Jaenisch R (2001) Synergism of Xist RNA, DNA methylation, and histone hypoacetylation in maintaining X chromosome inactivation. J Cell Biol 153: 773–784. doi: 10.1083/jcb.153.4.773
|
| [8] |
Li E, Beard C, Jaenisch R (1993) Role for DNA methylation in genomic imprinting. Nature 366: 362–365. doi: 10.1038/366362a0
|
| [9] |
Okano M, Bell DW, Haber DA, et al. (1999) DNA methyltransferases Dnmt3a and Dnmt3b are essential for de novo methylation and mammalian development. Cell 99: 247–257. doi: 10.1016/S0092-8674(00)81656-6
|
| [10] |
Hashimoto H, Liu Y, Upadhyay AK, et al. (2012) Recognition and potential mechanisms for replication and erasure of cytosine hydroxymethylation. Nucleic Acids Res 40: 4841–4849. doi: 10.1093/nar/gks155
|
| [11] |
Nabel CS, Jia H, Ye Y, et al. (2012) AID/APOBEC deaminases disfavor modified cytosines implicated in DNA demethylation. Nat Chem Biol 8: 751–758. doi: 10.1038/nchembio.1042
|
| [12] |
Bochtler M, Kolano A, Xu GL (2017) DNA demethylation pathways: Additional players and regulators. Bioessays 39: 1–13. doi: 10.1002/bies.201670013
|
| [13] |
Tahiliani M, Koh KP, Shen Y, et al. (2009) Conversion of 5-methylcytosine to 5-hydroxymethylcytosine in mammalian DNA by MLL partner TET1. Science 324: 930–935. doi: 10.1126/science.1170116
|
| [14] |
Tamanaha E, Guan S, Marks K, et al. (2016) Distributive processing by the iron(II)/α-ketoglutarate-dependent catalytic domains of the TET enzymes is consistent with epigenetic roles for oxidized 5-methylcytosine bases. J Am Chem Soc 138: 9345–9348. doi: 10.1021/jacs.6b03243
|
| [15] |
Müller U, Bauer C, Siegl M, et al. (2014) TET-mediated oxidation of methylcytosine causes TDG or NEIL glycosylase dependent gene reactivation. Nucleic Acids Res 42: 8592–8604. doi: 10.1093/nar/gku552
|
| [16] |
Weber AR, Krawczyk C, Robertson AB, et al. (2016) Biochemical reconstitution of TET1-TDG-BER-dependent active DNA demethylation reveals a highly coordinated mechanism. Nat Commun 7: 10806. doi: 10.1038/ncomms10806
|
| [17] |
Wang J, Hevi S, Kurash JK, et al. (2009) The lysine demethylase LSD1 (KDM1) is required for maintenance of global DNA methylation. Nat Genet 41: 125–129. doi: 10.1038/ng.268
|
| [18] |
Szwagierczak A, Bultmann S, Schmidt CS, et al. (2010) Sensitive enzymatic quantification of 5-hydroxymethylcytosine in genomic DNA. Nucleic Acids Res 38: e181. doi: 10.1093/nar/gkq684
|
| [19] |
Spruijt CG, Gnerlich F, Smits AH, et al. (2013) Dynamic readers for 5-(hydroxy)methylcytosine and its oxidized derivatives. Cell 152: 1146–1159. doi: 10.1016/j.cell.2013.02.004
|
| [20] | Matarese F, Carrillode SPE, Stunnenberg HG (2011) 5-Hydroxymethylcytosine: A new kid on the epigenetic block? Mol Syst Biol 7: 562. |
| [21] |
Koh KP, Yabuuchi A, Rao S, et al. (2011) Tet1 and Tet2 regulate 5-hydroxymethylcytosine production and cell lineage specification in mouse embryonic stem cells. Cell Stem Cell 8: 200–213. doi: 10.1016/j.stem.2011.01.008
|
| [22] |
Ito S, D'Alessio AC, Taranova OV, et al. (2010) Role of Tet proteins in 5mC to 5hmC conversion, ES-cell self-renewal and inner cell mass specification. Nature 466: 1129–1133. doi: 10.1038/nature09303
|
| [23] |
Caron G, Hussein M, Kulis M, et al. (2015) Cell-cycle-dependent reconfiguration of the DNA methylome during terminal differentiation of human B cells into plasma cells. Cell Rep 13: 1059–1071. doi: 10.1016/j.celrep.2015.09.051
|
| [24] |
Costa Y, Ding J, Theunissen TW, et al. (2013) NANOG-dependent function of TET1 and TET2 in establishment of pluripotency. Nature 495: 370–374. doi: 10.1038/nature11925
|
| [25] |
Kriaucionis S, Heintz N (2009) The nuclear DNA base 5-hydroxymethylcytosine is present in Purkinje neurons and the brain. Science 324: 929–930. doi: 10.1126/science.1169786
|
| [26] |
Jin SG, Wu X, Li AX, et al. (2011) Genomic mapping of 5-hydroxymethylcytosine in the human brain. Nucleic Acids Res 39: 5015–5024. doi: 10.1093/nar/gkr120
|
| [27] |
Nestor CE, Ottaviano R, Reddington J, et al. (2012) Tissue type is a major modifier of the 5-hydroxymethylcytosine content of human genes. Genome Res 22: 467–477. doi: 10.1101/gr.126417.111
|
| [28] |
Szulwach KE, Li X, Li Y, et al. (2011) 5-hmC-mediated epigenetic dynamics during postnatal neurodevelopment and aging. Nat Neurosci 14: 1607–1616. doi: 10.1038/nn.2959
|
| [29] |
Sherwani SI, Khan HA (2015) Role of 5-hydroxymethylcytosine in neurodegeneration. Gene 570: 17–24. doi: 10.1016/j.gene.2015.06.052
|
| [30] |
Jeschke J, Collignon E, Fuks F (2016) Portraits of TET-mediated DNA hydroxymethylation in cancer. Curr Opin Genet Dev 36: 16–26. doi: 10.1016/j.gde.2016.01.004
|
| [31] | Haffner MC, Chaux A, Meeker AK, et al. (2011) Global 5-hydroxymethylcytosine content is significantly reduced in tissue stem/progenitor cell compartments and in human cancers. Oncotarget 2: 627–637. |
| [32] |
Mahé EA, Madigou T, Sérandour AA, et al. (2017) Cytosine modifications modulate the chromatin architecture of transcriptional enhancers. Genome Res 27: 947–958. doi: 10.1101/gr.211466.116
|
| [33] |
Nan X, Ng HH, Johnson CA, et al. (1998) Transcriptional repression by the methyl-CpG-binding protein MeCP2 involves a histone deacetylase complex. Nature 393: 386–389. doi: 10.1038/30764
|
| [34] |
Ng HH, Bird A (1999) DNA methylation and chromatin modification. Curr Opin Genet Dev 9: 158–163. doi: 10.1016/S0959-437X(99)80024-0
|
| [35] |
Schübeler D (2015) Function and information content of DNA methylation. Nature 517: 321–326. doi: 10.1038/nature14192
|
| [36] |
Williams K, Christensen J, Pedersen MT, et al. (2011) TET1 and hydroxymethylcytosine in transcription and DNA methylation fidelity. Nature 473: 343–348. doi: 10.1038/nature10066
|
| [37] |
Sérandour AA, Avner S, Oger F, et al. (2012) Dynamic hydroxymethylation of deoxyribonucleic acid marks differentiation-associated enhancers. Nucleic Acids Res 40: 8255–8265. doi: 10.1093/nar/gks595
|
| [38] |
Song CX, He C (2013) Potential functional roles of DNA demethylation intermediates. Trends Biochem Sci 38: 480–484. doi: 10.1016/j.tibs.2013.07.003
|
| [39] | Sepulveda H, Villagra A, Montecino M (2017) Tet-mediated DNA demethylation is required for SWI/SNF-dependent chromatin remodeling and histone-modifying activities that trigger expression of the Sp7 osteoblast master gene during mesenchymal lineage commitment. Mol Cell Biol 37. |
| [40] |
Yildirim O, Li R, Hung JH, et al. (2011) Mbd3/NURD complex regulates expression of 5-hydroxymethylcytosine marked genes in embryonic stem cells. Cell 147: 1498–1510. doi: 10.1016/j.cell.2011.11.054
|
| [41] |
Neri F, Incarnato D, Krepelova A, et al. (2013) Genome-wide analysis identifies a functional association of Tet1 and Polycomb repressive complex 2 in mouse embryonic stem cells. Genome Biol 14: R91. doi: 10.1186/gb-2013-14-8-r91
|
| [42] |
Deplus R, Delatte B, Schwinn MK, et al. (2013) TET2 and TET3 regulate GlcNAcylation and H3K4 methylation through OGT and SET1/COMPASS. EMBO J 32: 645–655. doi: 10.1038/emboj.2012.357
|
| [43] |
Kong L, Tan L, Lv R, et al. (2016) A primary role of TET proteins in establishment and maintenance of De Novo bivalency at CpG islands. Nucleic Acids Res 44: 8682–8692. doi: 10.1093/nar/gkw529
|
| [44] | Mendonca A, Chang EH, Liu W, et al. (2014) Hydroxymethylation of DNA influences nucleosomal conformation and stability in vitro. BBA-Gene Regul Mech 1839: 1323–1329. |
| [45] |
Deplus R, Delatte B, Schwinn MK, et al. (2013) TET2 and TET3 regulate GlcNAcylation and H3K4 methylation through OGT and SET1/COMPASS. EMBO J 32: 645–655. doi: 10.1038/emboj.2012.357
|
| [46] |
Guan W, Guyot R, Samarut J, et al. (2017) Methylcytosine dioxygenase TET3 interacts with thyroid hormone nuclear receptors and stabilizes their association to chromatin. P Natl Acad Sci USA 114: 8229–8234. doi: 10.1073/pnas.1702192114
|
| [47] |
Zhang YW, Wang Z, Xie W, et al. (2017) Acetylation enhances TET2 function in protecting against abnormal DNA methylation during oxidative stress. Mol Cell 65: 323–335. doi: 10.1016/j.molcel.2016.12.013
|
| [48] |
Cimmino L, Dawlaty MM, Ndiaye-Lobry D, et al. (2015) TET1 is a tumor suppressor of hematopoietic malignancy. Nat Immunol 16: 653–662. doi: 10.1038/ni.3148
|
| [49] |
An J, González-Avalos E, Chawla A, et al. (2015) Acute loss of TET function results in aggressive myeloid cancer in mice. Nat Commun 6: 10071. doi: 10.1038/ncomms10071
|
| [50] |
Kafer GR, Li X, Horii T, et al. (2016) 5-Hydroxymethylcytosine marks sites of DNA damage and promotes genome stability. Cell Rep 14: 1283–1292. doi: 10.1016/j.celrep.2016.01.035
|
| [51] |
Mahfoudhi E, Talhaoui I, Cabagnols X, et al. (2016) TET2-mediated 5-hydroxymethylcytosine induces genetic instability and mutagenesis. DNA Rep 43: 78–88. doi: 10.1016/j.dnarep.2016.05.031
|
| [52] |
Jiang D, Zhang Y, Hart RP, et al. (2015) Alteration in 5-hydroxymethylcytosine-mediated epigenetic regulation leads to Purkinje cell vulnerability in ATM deficiency. Brain 138: 3520–3536. doi: 10.1093/brain/awv284
|
| [53] |
Jiang D, Wei S, Chen F, et al. (2017) TET3-mediated DNA oxidation promotes ATR-dependent DNA damage response. EMBO Rep 18: 781–796. doi: 10.15252/embr.201643179
|
| [54] |
Blackford AN, Jackson SP (2017) ATM, ATR, and DNA-PK: the trinity at the heart of the DNA damage response. Mol Cell 66: 801–817. doi: 10.1016/j.molcel.2017.05.015
|
| [55] |
Sellou H, Lebeaupin T, Chapuis C, et al. (2016) The poly(ADP-ribose)-dependent chromatin remodeler Alc1 induces local chromatin relaxation upon DNA damage. Mol Biol Cell 27: 3791–3799. doi: 10.1091/mbc.e16-05-0269
|
| [56] | Smith R, Sellou H, Chapuis C, et al. (2018) CHD3 and CHD4 recruitment and chromatin remodeling activity at DNA breaks is promoted by early poly(ADP-ribose)-dependent chromatin relaxation. Nucleic Acids Res 46: 6087. |
| [57] | Ciccarone F, Valentini E, Zampieri M, et al. (2015) 5mC-hydroxylase activity is influenced by the PARylation of TET1 enzyme. Oncotarget 6: 24333–24347. |
| [58] | Ciccarone F, Valentini E, Bacalini MG, et al. (2014) Poly(ADP-ribosyl)ation is involved in the epigenetic control of TET1 gene transcription. Oncotarget 5: 10356–10367. |
| [59] |
Chou DM, Adamson B, Dephoure NE, et al. (2010) A chromatin localization screen reveals poly (ADP ribose)-regulated recruitment of the repressive polycomb and NuRD complexes to sites of DNA damage. P Natl Acad Sci USA 107: 18475–18480. doi: 10.1073/pnas.1012946107
|
| [60] |
Luijsterburg MS, Dinant C, Lans H, et al. (2009) Heterochromatin protein 1 is recruited to various types of DNA damage. J Cell Biol 185: 577–586. doi: 10.1083/jcb.200810035
|
| [61] | Abu-Zhayia ER, Awwad SW, Ben-Oz B, et al. (2017) CDYL1 fosters double-strand break-induced transcription silencing and promotes homology-directed repair. J Mol Cell Biol 1: 1. |
| [62] |
D'Alessandro G, Fagagna FDD (2017) Transcription and DNA damage: holding hands or crossing swords? J Mol Biol 429: 3215–3229. doi: 10.1016/j.jmb.2016.11.002
|
| [63] | Puc J, Aggarwal AK, Rosenfeld MG (2017) Physiological functions of programmed DNA breaks in signal-induced transcription. Nat Rev Mol Cell Bio 18: 471–476. |
| [64] |
Ju BG, Lunyak VV, Perissi V, et al. (2006) A topoisomerase IIbeta-mediated dsDNA break required for regulated transcription. Science 312: 1798–1802. doi: 10.1126/science.1127196
|
| [65] |
Madabhushi R, Gao F, Pfenning AR, et al. (2015) Activity-induced DNA breaks govern the expression of neuronal early-response genes. Cell 161: 1592–1605. doi: 10.1016/j.cell.2015.05.032
|
| [66] |
Perillo B, Ombra MN, Bertoni A, et al. (2008) DNA oxidation as triggered by H3K9me2 demethylation drives estrogen-induced gene expression. Science 319: 202–206. doi: 10.1126/science.1147674
|
| [67] |
Puc J, Kozbial P, Li W, et al. (2015) Ligand-dependent enhancer activation regulated by topoisomerase-I activity. Cell 160: 367–380. doi: 10.1016/j.cell.2014.12.023
|
| [68] |
Baranello L, Wojtowicz D, Cui K, et al. (2016) RNA polymerase II regulates topoisomerase 1 activity to favor efficient transcription. Cell 165: 357–371. doi: 10.1016/j.cell.2016.02.036
|
| [69] |
Bunch H, Lawney BP, Lin YF, et al. (2015) Transcriptional elongation requires DNA break-induced signalling. Nat Commun 6: 10191. doi: 10.1038/ncomms10191
|
| [70] |
Marnef A, Cohen S, Legube G (2017) Transcription-coupled DNA double-strand break repair: active genes need special care. J Mol Biol 429: 1277–1288. doi: 10.1016/j.jmb.2017.03.024
|
| [71] |
Huertas P, Aguilera A (2003) Cotranscriptionally formed DNA:RNA hybrids mediate transcription elongation impairment and transcription-associated recombination. Mol Cell 12: 711–721. doi: 10.1016/j.molcel.2003.08.010
|
| [72] |
Sollier J, Stork CT, García-Rubio ML, et al. (2014) Transcription-coupled nucleotide excision repair factors promote R-loop-induced genome instability. Mol Cell 56: 777–785. doi: 10.1016/j.molcel.2014.10.020
|
| [73] |
Métivier R, Gallais R, Tiffoche C, et al. (2008) Cyclical DNA methylation of a transcriptionally active promoter. Nature 452: 45–50. doi: 10.1038/nature06544
|
| [74] |
Li J, Wu X, Zhou Y, et al. (2018) Decoding the dynamic DNA methylation and hydroxymethylation landscapes in endodermal lineage intermediates during pancreatic differentiation of hESC. Nucleic Acids Res 46: 2883–2900. doi: 10.1093/nar/gky063
|
| [75] |
Zhang Y, Zhang D, Li Q, et al. (2016) Nucleation of DNA repair factors by FOXA1 links DNA demethylation to transcriptional pioneering. Nat Genet 48: 1003–1013. doi: 10.1038/ng.3635
|
| [76] |
Boque-Sastre R, Soler M, Oliveira-Mateos C, et al. (2015) Head-to-head antisense transcription and R-loop formation promotes transcriptional activation. P Natl Acad Sci USA 112: 5785–5790. doi: 10.1073/pnas.1421197112
|
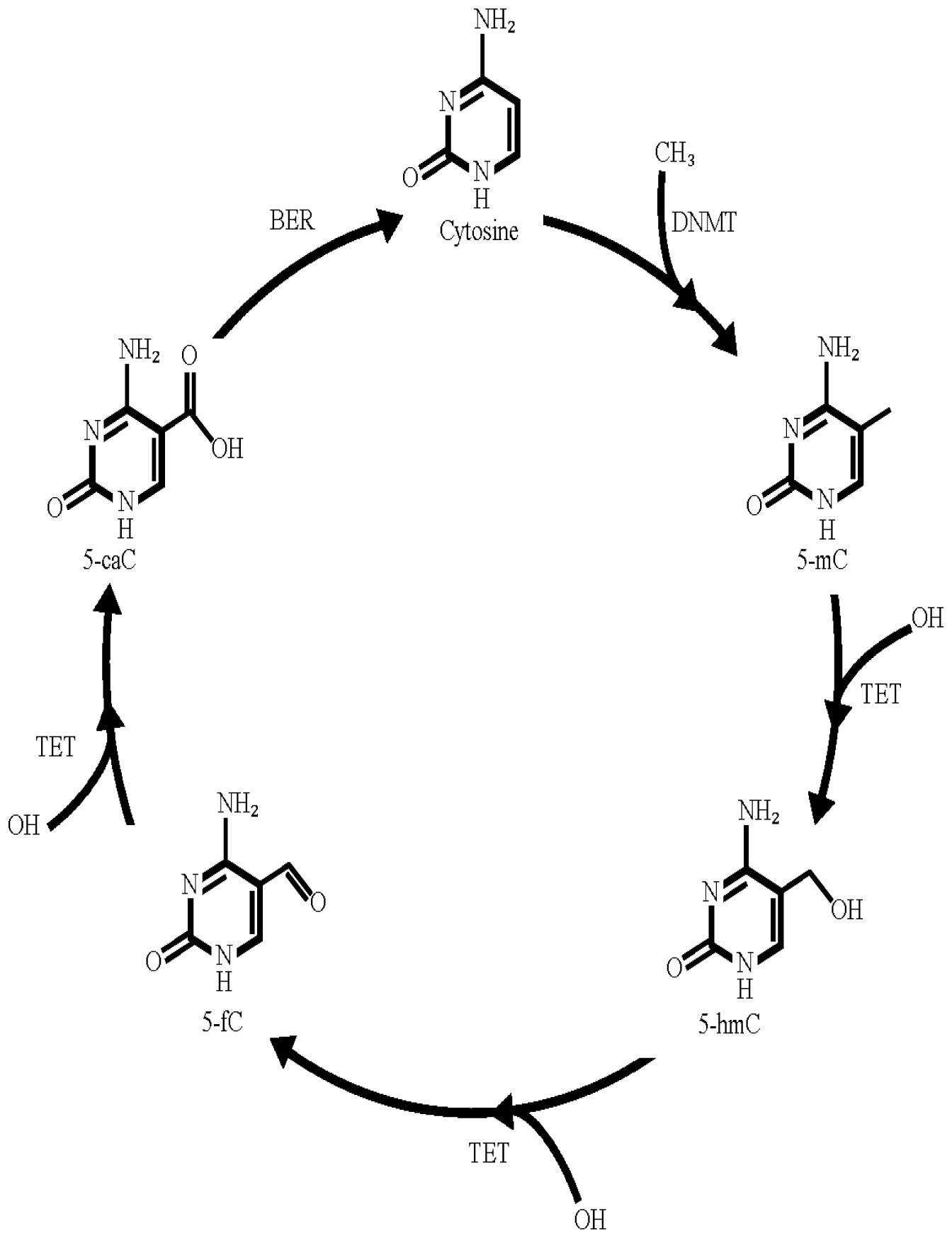
Figures(1)

Audrey Lejart, Gilles Salbert, Sébastien Huet. Cytosine hydroxymethylation by TET enzymes: From the control of gene expression to the regulation of DNA repair mechanisms, and back[J]. AIMS Biophysics, 2018, 5(3): 182-193. doi: 10.3934/biophy.2018.3.182







 DownLoad:
DownLoad: