Citation: Pravin D Potdar, Aashutosh U Shetti. Molecular Biomarkers for Diagnosis & Therapies of Alzheimer’s Disease[J]. AIMS Neuroscience, 2016, 3(4): 433-453. doi: 10.3934/Neuroscience.2016.4.433

| [1] |
Butterfield D, Castegna A, Lauderback C, et al. (2002) Evidence that amyloid beta-peptide-induced lipid peroxidation and its sequelae in Alzheimer’s disease brain contribute to neuronal death. Neurobiol Aging 23: 655-664. doi:10.1016/S0197-4580(01)00340-2. doi: 10.1016/S0197-4580(01)00340-2

|
| [2] |
Ramsden M, Kotilinek L, Forster C, et al. (2005) Age-dependent neurofibrillary tangle formation, neuron loss, and memory impairment in a mouse model of human tauopathy (P301L). J Neurosci 25: 10637-10647. doi:10.1523/JNEUROSCI.3279-05.2005. doi: 10.1523/JNEUROSCI.3279-05.2005
|
| [3] |
Serpell LC (2000) Alzheimer’s amyloid fibrils: structure and assembly. Biochim Biophys Acta- Mol Basis Dis 1502: 16-30. doi:10.1016/S0925-4439(00)00029-6. doi: 10.1016/S0925-4439(00)00029-6
|
| [4] |
Nunan J, Small DH (2000) Regulation of APP cleavage by α-, β- and γ-secretases. FEBS Lett 483: 6-10. doi:10.1016/S0014-5793(00)02076-7. doi: 10.1016/S0014-5793(00)02076-7
|
| [5] | Chasseigneaux S, Allinquant B (2012) Functions of Aβ, sAPPα and sAPPβ : similarities and differences. J Neurochem 120 Suppl: 99-108. doi:10.1111/j.1471-4159.2011.07584.x. |
| [6] |
Sadik G, Kaji H, Takeda K, et al. (1999) In vitro processing of amyloid precursor protein by cathepsin D. Int J Biochem Cell Biol 31: 1327-1337. http://www.ncbi.nlm.nih.gov/pubmed/10605825. Accessed January 3, 2016. doi: 10.1016/S1357-2725(99)00053-9
|
| [7] | Kume H, Maruyama K, Kametani F (2004) Intracellular domain generation of amyloid precursor protein by epsilon-cleavage depends on C-terminal fragment by alpha-secretase cleavage. Int J Mol Med 13: 121-125. http://www.ncbi.nlm.nih.gov/pubmed/14654982. |
| [8] |
Konietzko U (2012) AICD nuclear signaling and its possible contribution to Alzheimer’s disease. Curr Alzheimer Res 9: 200-216. http://www.ncbi.nlm.nih.gov/pubmed/21605035. doi: 10.2174/156720512799361673
|
| [9] | Brion JP, Couck AM, Passareiro E, et al. (1985) Neurofibrillary tangles of Alzheimer’s disease: an immunohistochemical study. J Submicrosc Cytol 17: 89-96. http://europepmc.org/abstract/med/3973960. |
| [10] |
Ferrer I, Gomez-Isla T, Puig B, et al. (2005) Current advances on different kinases involved in tau phosphorylation, and implications in Alzheimer’s disease and tauopathies. Curr Alzheimer Res 2: 3-18. http://www.ncbi.nlm.nih.gov/pubmed/15977985. doi: 10.2174/1567205052772713
|
| [11] |
Williams DR (2006) Tauopathies: classification and clinical update on neurodegenerative diseases associated with microtubule-associated protein tau. Intern Med J 36: 652-660. doi:10.1111/j.1445-5994.2006.01153.x. doi: 10.1111/j.1445-5994.2006.01153.x
|
| [12] |
Levy-Lahad E, Wasco W, Poorkaj P, et al. (1995) Candidate gene for the chromosome 1 familial Alzheimer’s disease locus. Science 269: 973-977. doi:10.1126/science.7638622. doi: 10.1126/science.7638622
|
| [13] |
Bertram L, Blacker D, Mullin K, et al. (2000) Evidence for genetic linkage of Alzheimer’s disease to chromosome 10q. Science 290: 2302-2303. doi:10.1126/science.290.5500.2302. doi: 10.1126/science.290.5500.2302
|
| [14] | Beecham GW, Martin ER, Li Y-J, et al. (2008) Genome-wide association study implicates a chromosome 12 risk locus for late-onset Alzheimer disease. Am J Hum Genet 84: 35-43. doi:10.1016/j.ajhg.2008.12.008. |
| [15] |
Wilkins CH, Sheline YI, Roe CM, et al. (2006) Vitamin D deficiency is associated with low mood and worse cognitive performance in older adults. Am J Geriatr Psychiatry 14: 1032-1040. doi:10.1097/01.JGP.0000240986.74642.7c. doi: 10.1097/01.JGP.0000240986.74642.7c
|
| [16] |
Mullan M, Houlden H, Windelspecht M, et al. (1992) A locus for familial early-onset Alzheimer’s disease on the long arm of chromosome 14, proximal to the alpha 1-antichymotrypsin gene. Nat Genet 2: 340-342. doi:10.1038/ng1292-340. doi: 10.1038/ng1292-340
|
| [17] |
St George-Hyslop P, Haines J, Rogaev E, et al. (1992) Genetic evidence for a novel familial Alzheimer’s disease locus on chromosome 14. Nat Genet 2: 330-334. doi:10.1038/ng1292-330. doi: 10.1038/ng1292-330
|
| [18] | Pericak-Vance M a, Bebout JL, Gaskell PC, et al. (1991) Linkage studies in familial Alzheimer disease: evidence for chromosome 19 linkage. Am J Hum Genet 48: 1034-1050. http://www.ncbi.nlm.nih.gov/pmc/articles/PMC1683100/pdf/ajhg00090-0019.pdf. |
| [19] |
Chartier-Harlin MC, Parfitt M, Legrain S, et al. (1994) Apolipoprotein E, epsilon 4 allele as a major risk factor for sporadic early and late-onset forms of Alzheimer’s disease: analysis of the 19q13.2 chromosomal region. Hum Mol Genet 3: 569-574. doi:10.1093/hmg/3.4.569. doi: 10.1093/hmg/3.4.569
|
| [20] |
Yu J-T, Tan L, Hardy J (2014) Apolipoprotein E in Alzheimer’s disease: an update. Annu Rev Neurosci 37: 79-100. doi:10.1146/annurev-neuro-071013-014300. doi: 10.1146/annurev-neuro-071013-014300
|
| [21] |
Strittmatter WJ, Weisgraber KH, Goedert M, et al. (1994) Hypothesis: microtubule instability and paired helical filament formation in the Alzheimer disease brain are related to apolipoprotein E genotype. Exp Neurol 125: 163-171. doi:S0014488684710193. doi: 10.1006/exnr.1994.1019
|
| [22] |
Wu L, Rosa-Neto P, Hsiung G-YR, et al. (2012) Early-onset familial Alzheimer’s disease (EOFAD). Can J Neurol Sci 39: 436-445. doi:W4438L6488727555. doi: 10.1017/S0317167100013949
|
| [23] |
Olsson A, Vanderstichele H, Andreasen N, et al. (2005) Simultaneous measurement of β-amyloid (1-42), total Tau, and phosphorylated Tau (Thr181) in cerebrospinal fluid by the xMAP technology. Clin Chem 51: 336-345. doi:10.1373/clinchem.2004.039347. doi: 10.1373/clinchem.2004.039347
|
| [24] |
Petraki CD, Karavana VN, Skoufogiannis PT, et al. (2001) The spectrum of human kallikrein 6 (zyme/protease M/neurosin) expression in human tissues as assessed by immunohistochemistry. J Histochem Cytochem 49: 1431-1441. http://www.ncbi.nlm.nih.gov/pubmed/11668196. doi: 10.1177/002215540104901111
|
| [25] |
Rockenstein E, Nuber S, Overk CR, et al. (2014) Accumulation of oligomer-prone α-synuclein exacerbates synaptic and neuronal degeneration in vivo. Brain 137: 1496-1513. doi:10.1093/brain/awu057. doi: 10.1093/brain/awu057
|
| [26] |
Spencer B, Valera E, Rockenstein E, et al. (2015) A brain-targeted, modified neurosin (kallikrein-6) reduces α-synuclein accumulation in a mouse model of multiple system atrophy. Mol Neurodegener 10: 48. doi:10.1186/s13024-015-0043-6. doi: 10.1186/s13024-015-0043-6
|
| [27] |
Ogawa K, Yamada T, Tsujioka Y, et al. (2000) Localization of a novel type trypsin-like serine protease, neurosin, in brain tissues of Alzheimer’s disease and Parkinson's disease. Psychiatry Clin Neurosci 54: 419-426. doi:10.1046/j.1440-1819.2000.00731.x. doi: 10.1046/j.1440-1819.2000.00731.x
|
| [28] |
Diamandis EP, Yousef GM, Petraki C, et al. (2000). Human kallikrein 6 as a biomarker of alzheimer’s disease. Clin Biochem 33:663-667. http://www.ncbi.nlm.nih.gov/pubmed/11166014. doi: 10.1016/S0009-9120(00)00185-5
|
| [29] |
Saper CB, Scammell TE, Lu J (2005) Hypothalamic regulation of sleep and circadian rhythms. Nature 437: 1257-1263. doi:10.1038/nature04284. doi: 10.1038/nature04284
|
| [30] |
de Lecea L, Kilduff TS, Peyron C, et al. (1998) The hypocretins: Hypothalamus-specific peptides with neuroexcitatory activity. Proc Natl Acad Sci 95: 322-327. doi:10.1073/pnas.95.1.322. doi: 10.1073/pnas.95.1.322
|
| [31] |
Liguori C, Romigi A, Nuccetelli M, et al. (2014) Orexinergic System Dysregulation, Sleep Impairment, and Cognitive Decline in Alzheimer Disease. JAMA Neurol 71: 1498-1505. doi:10.1001/jamaneurol.2014.2510. doi: 10.1001/jamaneurol.2014.2510
|
| [32] |
Baumann CR, Hersberger M, Bassetti CL (2006) Hypocretin-1 (orexin A) levels are normal in Huntington’s disease. J Neurol 253: 1232-1233. doi:10.1007/s00415-006-0146-7. doi: 10.1007/s00415-006-0146-7
|
| [33] | Dauvilliers YA, Lehmann S, Jaussent I, et al. (2014). Hypocretin and brain β-amyloid peptide interactions in cognitive disorders and narcolepsy. Front Aging Neurosci 6. doi:10.3389/fnagi.2014.00119. |
| [34] |
Friedman LF, Zeitzer JM, Lin L, et al. et al. (2007) In Alzheimer disease, increased wake fragmentation found in those with lower hypocretin-1. Neurology 68: 793-794. doi:10.1212/01.wnl.0000256731.57544.f9. doi: 10.1212/01.wnl.0000256731.57544.f9
|
| [35] |
Slats D, Claassen JAHR, Lammers GJ, et al. (2012) Association between hypocretin-1 and amyloid-β42 cerebrospinal fluid levels in Alzheimer’s disease and healthy controls. Curr Alzheimer Res 9: 1119-1125. http://www.ncbi.nlm.nih.gov/pubmed/22742854. doi: 10.2174/156720512804142840
|
| [36] |
Fronczek R, van Geest S, Frölich M, et al. (2012) Hypocretin (orexin) loss in Alzheimer’s disease. Neurobiol Aging 33: 1642-1650. doi:10.1016/j.neurobiolaging.2011.03.014. doi: 10.1016/j.neurobiolaging.2011.03.014
|
| [37] |
Gallone S, Boschi S, Rubino E, et al. et al. (2014) Is HCRTR2 a genetic risk factor for Alzheimer’s disease? Dement Geriatr Cogn Disord 38: 245-253. doi:10.1159/000359964. doi: 10.1159/000359964
|
| [38] | Tucker HM, Kihiko M, Caldwell JN, et al. (2000) The plasmin system is induced by and degrades amyloid-beta aggregates. J Neurosci 20: 3937-3946. doi:20/11/3937. |
| [39] |
Man H-Y, Ma X-M (2012) A role for neuroserpin in neuron morphological development. J Neurochem 121: 495-496. doi:10.1111/j.1471-4159.2012.07655.x. doi: 10.1111/j.1471-4159.2012.07655.x
|
| [40] |
Ledesma MD, Da Silva JS, Crassaerts K, et al. (2000) Brain plasmin enhances APP alpha-cleavage and Abeta degradation and is reduced in Alzheimer’s disease brains. EMBO Rep 1: 530-535. doi:10.1093/embo-reports/kvd107. doi: 10.1093/embo-reports/kvd107
|
| [41] | Hanzel CE, Iulita MF, Eyjolfsdottir H, et al. (2014) Analysis of matrix metallo-proteases and the plasminogen system in mild cognitive impairment and Alzheimer’s disease cerebrospinal fluid. J Alzheimers Dis 40: 667-678. doi:10.3233/JAD-132282. |
| [42] |
Kinghorn KJ, Crowther DC, Sharp LK, et al. (2006) Neuroserpin binds Abeta and is a neuroprotective component of amyloid plaques in Alzheimer disease. J Biol Chem 281: 29268-29277. doi:10.1074/jbc.M600690200. doi: 10.1074/jbc.M600690200
|
| [43] |
Fabbro S, Schaller K, Seeds NW (2011) Amyloid-beta levels are significantly reduced and spatial memory defects are rescued in a novel neuroserpin-deficient Alzheimer’s disease transgenic mouse model. J Neurochem 118: 928-938. doi:10.1111/j.1471-4159.2011.07359.x. doi: 10.1111/j.1471-4159.2011.07359.x
|
| [44] |
Subhadra B, Schaller K, Seeds NW (2013) Neuroserpin up-regulation in the Alzheimer’s disease brain is associated with elevated thyroid hormone receptor-β1 and HuD expression. Neurochem Int 63: 476-481. doi:10.1016/j.neuint.2013.08.010. doi: 10.1016/j.neuint.2013.08.010
|
| [45] |
Okada T, Kajimoto T, Jahangeer S, et al. (2009) Sphingosine kinase/sphingosine 1-phosphate signalling in central nervous system. Cell Signal 21: 7-13. doi:10.1016/j.cellsig.2008.07.011. doi: 10.1016/j.cellsig.2008.07.011
|
| [46] |
Cieślik M, Czapski GA, Strosznajder JB (2015) The Molecular Mechanism of Amyloid β42 Peptide Toxicity: The Role of Sphingosine Kinase-1 and Mitochondrial Sirtuins. PLoS One 10: e0137193. doi:10.1371/journal.pone.0137193. doi: 10.1371/journal.pone.0137193
|
| [47] |
Carro E, Trejo JL, Gomez-Isla T, et al. (2002) Serum insulin-like growth factor I regulates brain amyloid-beta levels. Nat Med 8: 1390-1397. doi:10.1038/nm793. doi: 10.1038/nm1202-793
|
| [48] | Mizugishi K, Yamashita T, Olivera A, et al. (2005) Essential role for sphingosine kinases in neural and vascular development. Mol Cell Biol 2511113-11121. doi:10.1128/MCB.25.24.11113-11121.2005. |
| [49] |
Couttas T a, Kain N, Daniels B, et al. (2014) Loss of the neuroprotective factor Sphingosine 1-phosphate early in Alzheimer’s disease pathogenesis. Acta Neuropathol Commun 2: 9. doi:10.1186/2051-5960-2-9. doi: 10.1186/2051-5960-2-9
|
| [50] |
Sivasubramanian M, Kanagaraj N, Dheen ST, et al. (2015) Sphingosine kinase 2 and sphingosine-1-phosphate promotes mitochondrial function in dopaminergic neurons of mouse model of Parkinson’s disease and in MPP+-treated MN9D cells in vitro. Neuroscience 290: 636-648. doi:10.1016/j.neuroscience.2015.01.032. doi: 10.1016/j.neuroscience.2015.01.032
|
| [51] |
Takasugi N, Sasaki T, Suzuki K, et al. (2011) BACE1 activity is modulated by cell-associated sphingosine-1-phosphate. J Neurosci 31: 6850-6857. doi:10.1523/JNEUROSCI.6467-10. doi: 10.1523/JNEUROSCI.6467-10.2011
|
| [52] |
Hagen N, Hans M, Hartmann D, et al. (2011) Sphingosine-1-phosphate links glycosphingolipid metabolism to neurodegeneration via a calpain-mediated mechanism. Cell Death Differ 18: 1356-1365. doi:10.1038/cdd.2011.7. doi: 10.1038/cdd.2011.7
|
| [53] |
Huh CG, Håkansson K, Nathanson CM, et al. (1999) Decreased metastatic spread in mice homozygous for a null allele of the cystatin C protease inhibitor gene. Mol Pathol 52: 332-340. http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=395718&tool=pmcentrez&rendertype=abstract. doi: 10.1136/mp.52.6.332
|
| [54] |
Wilson ME, Boumaza I, Lacomis D, et al. (2010) Cystatin C: a candidate biomarker for amyotrophic lateral sclerosis. PLoS One 5: e15133. doi:10.1371/journal.pone.0015133. doi: 10.1371/journal.pone.0015133
|
| [55] |
Simonsen AH, McGuire J, Podust VN, et al. (2007) A novel panel of cerebrospinal fluid biomarkers for the differential diagnosis of Alzheimer’s disease versus normal aging and frontotemporal dementia. Dement Geriatr Cogn Disord 24: 434-440. doi:10.1159/000110576. doi: 10.1159/000110576
|
| [56] | Chen J, Liu C (2014) Association Of Serum Cystatin C Levels On The Progression And Cognition In Parkinson’s Disease (P4.045). Neurology 82: 45. |
| [57] |
Chuo L-J, Sheu WHH, Pai M-C, et al. (2007) Genotype and plasma concentration of cystatin C in patients with late-onset Alzheimer disease. Dement Geriatr Cogn Disord 23: 251-257. doi:10.1159/000100021. doi: 10.1159/000100021
|
| [58] |
Crawford FC, Freeman MJ, Schinka JA, et al. (2000) A polymorphism in the cystatin C gene is a novel risk factor for late-onset Alzheimer’s disease. Neurology 55: 763-768. http://www.ncbi.nlm.nih.gov/pubmed/10993992. doi: 10.1212/WNL.55.6.763
|
| [59] |
Ghidoni R, Paterlini A, Albertini V, et al. (2011) Cystatin C is released in association with exosomes: a new tool of neuronal communication which is unbalanced in Alzheimer’s disease. Neurobiol Aging 32: 1435-1442. doi:10.1016/j.neurobiolaging.2009.08.013. doi: 10.1016/j.neurobiolaging.2009.08.013
|
| [60] | Afonso S, Romagnano L, Babiarz B (2016) The expression and function of cystatin C and cathepsin B and cathepsin L during mouse embryo implantation and placentation. Development 124: 3415-3425. http://www.ncbi.nlm.nih.gov/pubmed/9310336. |
| [61] |
Sastre M, Calero M, Pawlik M, et al. (2004) Binding of cystatin C to Alzheimer’s amyloid beta inhibits in vitro amyloid fibril formation. Neurobiol Aging 25: 1033-1043. doi:10.1016/j.neurobiolaging.2003.11.006. doi: 10.1016/j.neurobiolaging.2003.11.006
|
| [62] |
Maruyama K, Ikeda S, Ishihara T, et al. (1990) Immunohistochemical characterization of cerebrovascular amyloid in 46 autopsied cases using antibodies to beta protein and cystatin C. Stroke 21: 397-403. doi:10.1161/01.STR.21.3.397. doi: 10.1161/01.STR.21.3.397
|
| [63] |
Deng A, Irizarry MC, Nitsch RM, et al. (2001). Elevation of cystatin C in susceptible neurons in Alzheimer’s disease. Am J Pathol 159: 1061-1068. doi:10.1016/S0002-9440(10)61781-6. doi: 10.1016/S0002-9440(10)61781-6
|
| [64] |
Ponomareva OY, Holmen IC, Sperry AJ, et al. (2014) Calsyntenin-1 regulates axon branching and endosomal trafficking during sensory neuron development in vivo. J Neurosci 34: 9235-9248. doi:10.1523/JNEUROSCI.0561-14.2014. doi: 10.1523/JNEUROSCI.0561-14.2014
|
| [65] |
Vagnoni A, Perkinton MS, Gray EH, et al. (2012) Calsyntenin-1 mediates axonal transport of the amyloid precursor protein and regulates Aβ production. Hum Mol Genet 21: 2845-2854. doi:10.1093/hmg/dds109. doi: 10.1093/hmg/dds109
|
| [66] |
Yin GN, Lee HW, Cho J-Y, et al. (2009) Neuronal pentraxin receptor in cerebrospinal fluid as a potential biomarker for neurodegenerative diseases. Brain Res 1265: 158-170. doi:10.1016/j.brainres.2009.01.058. doi: 10.1016/j.brainres.2009.01.058
|
| [67] |
Ludwig A, Blume J, Diep T-M, et al. (2009) Calsyntenins mediate TGN exit of APP in a kinesin-1-dependent manner. Traffic 10: 572-589. doi:10.1111/j.1600-0854.2009.00886.x. doi: 10.1111/j.1600-0854.2009.00886.x
|
| [68] |
Pettem KL, Yokomaku D, Luo L, et al. (2013) The specific α-neurexin interactor calsyntenin-3 promotes excitatory and inhibitory synapse development. Neuron 80: 113-128. doi:10.1016/j.neuron.2013.07.016. doi: 10.1016/j.neuron.2013.07.016
|
| [69] |
Uchida Y, Gomi F, Murayama S, et al. (2013) Calsyntenin-3 C-terminal fragment accumulates in dystrophic neurites surrounding aβ plaques in tg2576 mouse and Alzheimer disease brains: its neurotoxic role in mediating dystrophic neurite formation. Am J Pathol 182: 1718-1726. doi:10.1016/j.ajpath.2013.01.014. doi: 10.1016/j.ajpath.2013.01.014
|
| [70] |
DeSilva U, D’Arcangelo G, Braden VV, et al. (1997) The human reelin gene: Isolation, sequencing, and mapping on chromosome 7. Genome Res 7: 157-164. doi:10.1101/gr.7.2.157. doi: 10.1101/gr.7.2.157
|
| [71] |
Stranahan AM, Erion JR, Wosiski-Kuhn M (2013) Reelin signaling in development, maintenance, and plasticity of neural networks. Ageing Res Rev 12: 815-822. doi:10.1016/j.arr.2013.01.005. doi: 10.1016/j.arr.2013.01.005
|
| [72] |
Eastwood SL, Harrison PJ (2003) Interstitial white matter neurons express less reelin and are abnormally distributed in schizophrenia: towards an integration of molecular and morphologic aspects of the neurodevelopmental hypothesis. Mol Psychiatry 8: 821-831. doi:10.1038/sj.mp.4001399. doi: 10.1038/sj.mp.4001371
|
| [73] |
Fatemi SH, Stary JM, Egan EA (2002) Reduced blood levels of reelin as a vulnerability factor in pathophysiology of autistic disorder. Cell Mol Neurobiol 22: 139-152. doi:http://dx.doi.org/10.1023/A:1019857620251. doi: 10.1023/A:1019857620251
|
| [74] |
Botella-López A, Burgaya F, Gavín R, et al. (2006) Reelin expression and glycosylation patterns are altered in Alzheimer’s disease. Proc Natl Acad Sci U S A 103: 5573-5578. doi:10.1073/pnas.0601279103. doi: 10.1073/pnas.0601279103
|
| [75] | Herring A, Donath A, Steiner KM, et al. (2012) Reelin depletion is an early phenomenon of alzheimer’s pathology. J Alzheimer’s Dis 30: 963-979. doi:10.3233/JAD-2012-112069. |
| [76] | Chin J, Massaro CM, Palop JJ, et al. (2007) Reelin Depletion in the Entorhinal Cortex of Human Amyloid Precursor Protein Transgenic Mice and Humans with Alzheimer’s Disease. J Neurosci 27. |
| [77] | Pujadas L, Rossi D, Andrés R, et al. (2014) Reelin delays amyloid-beta fibril formation and rescues cognitive deficits in a model of Alzheimer’s disease. Nat Commun 5: 3443. doi:10.1038/ncomms4443. |
| [78] | Cuchillo-Ibáñez I, Balmaceda V, Botella-López A, et al. (2013) Beta-Amyloid Impairs Reelin Signaling. PLoS One 8: 1-10. doi:10.1371/journal.pone.0072297. |
| [79] |
Patrick GN, Zukerberg L, Nikolic M, et al. (1999) Conversion of p35 to p25 deregulates Cdk5 activity and promotes neurodegeneration. Nature 402: 615-622. doi:10.1038/45159. doi: 10.1038/45159
|
| [80] |
Sakamuro D, Elliott KJ, Wechsler-Reya R, et al. (1996) BIN1 is a novel MYC-interacting protein with features of a tumour suppressor. Nat Genet 14: 69-77. doi:10.1038/ng0996-69. doi: 10.1038/ng0996-69
|
| [81] |
Chapuis J, Hansmannel F, Gistelinck M, et al. (2013) Increased expression of BIN1 mediates Alzheimer genetic risk by modulating tau pathology. Mol Psychiatry 18: 1225-1234. doi:10.1038/mp.2013.1. doi: 10.1038/mp.2013.1
|
| [82] | Holler CJ, Davis PR, Beckett TL, et al. (2014) Bridging integrator 1 (BIN1) protein expression increases in the Alzheimer’s disease brain and correlates with neurofibrillary tangle pathology. J Alzheimers Dis 42: 1221-1227. doi:10.3233/JAD-132450. |
| [83] | Sun L, Tan M-S, Hu N, et al. (2013) Exploring the value of plasma BIN1 as a potential biomarker for alzheimer’s disease. J Alzheimers Dis 37: 291-295. doi:10.3233/JAD-130392. |
| [84] | Glennon EBC, Whitehouse IJ, Miners JS, et al. (2013) BIN1 Is Decreased in Sporadic but Not Familial Alzheimer’s Disease or in Aging. PLoS One 8. doi:10.1371/journal.pone.0078806. |
| [85] |
Gan-Or Z, Amshalom I, Bar-Shira A, et al. (2015) The Alzheimer disease BIN1 locus as a modifier of GBA-associated Parkinson disease. J Neurol 262: 2443-2447. doi:10.1007/s00415-015-7868-3. doi: 10.1007/s00415-015-7868-3
|
| [86] | Ou SH, Wu F, Harrich D, et al. (1995) Cloning and characterization of a novel cellular protein, TDP-43, that binds to human immunodeficiency virus type 1 TAR DNA sequence motifs. J Virol 69: 3584-3596. http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=189073&tool=pmcentrez&rendertype=abstract. |
| [87] |
Arai T, Mackenzie IRA, Hasegawa M, et al. (2009) Phosphorylated TDP-43 in Alzheimer’s disease and dementia with Lewy bodies. Acta Neuropathol 117: 125-136. doi:10.1007/s00401-008-0480-1. doi: 10.1007/s00401-008-0480-1
|
| [88] |
Foulds P, McAuley E, Gibbons L, et al. (2008) TDP-43 protein in plasma may index TDP-43 brain pathology in Alzheimer’s disease and frontotemporal lobar degeneration. Acta Neuropathol 116: 141-146. doi:10.1007/s00401-008-0389-8. doi: 10.1007/s00401-008-0389-8
|
| [89] |
Higashi S, Iseki E, Yamamoto R, et al. (2007) Concurrence of TDP-43, tau and alpha-synuclein pathology in brains of Alzheimer’s disease and dementia with Lewy bodies. Brain Res 1184: 284-294. doi:10.1016/j.brainres.2007.09.048. doi: 10.1016/j.brainres.2007.09.048
|
| [90] |
Zhang Y-J, Xu Y-F, Cook C, et al. (2009) Aberrant cleavage of TDP-43 enhances aggregation and cellular toxicity. Proc Natl Acad Sci U S A 106: 7607-7612. doi:10.1073/pnas.0900688106. doi: 10.1073/pnas.0900688106
|
| [91] |
Tauffenberger A, Chitramuthu BP, Bateman A, et al. (2013) Reduction of polyglutamine toxicity by TDP-43, FUS and progranulin in Huntington’s disease models. Hum Mol Genet 22: 782-794. doi:10.1093/hmg/dds485. doi: 10.1093/hmg/dds485
|
| [92] |
Josephs KA, Whitwell JL, Weigand SD, et al. (2014) TDP-43 is a key player in the clinical features associated with Alzheimer’s disease. Acta Neuropathol 127: 811-824. doi:10.1007/s00401-014-1269-z. doi: 10.1007/s00401-014-1269-z
|
| [93] |
Spilker C, Braunewell K-H (2003) Calcium-myristoyl switch, subcellular localization, and calcium-dependent translocation of the neuronal calcium sensor protein VILIP-3, and comparison with VILIP-1 in hippocampal neurons. Mol Cell Neurosci 24: 766-778. http://www.ncbi.nlm.nih.gov/pubmed/14664824. Accessed February 15, 2016. doi: 10.1016/S1044-7431(03)00242-2
|
| [94] |
Laterza OF, Modur VR, Crimmins DL, et al. (2006) Identification of novel brain biomarkers. Clin Chem 52: 1713-1721. doi:10.1373/clinchem.2006.070912. doi: 10.1373/clinchem.2006.070912
|
| [95] | Braunewell KH (2012) The visinin-like proteins VILIP-1 and VILIP-3 in Alzheimer’s disease-old wine in new bottles. Front Mol Neurosci 5: 20. doi:10.3389/fnmol.2012.00020. |
| [96] |
Chakroborty S, Stutzmann GE (2011) Early calcium dysregulation in Alzheimer’s disease: Setting the stage for synaptic dysfunction. Sci China Life Sci 54: 752-762. doi:10.1007/s11427-011-4205-7. doi: 10.1007/s11427-011-4205-7
|
| [97] |
Bezprozvanny I, Mattson MP (2008) Neuronal calcium mishandling and the pathogenesis of Alzheimer’s disease. Trends Neurosci 31: 454-463. doi:10.1016/j.tins.2008.06.005. doi: 10.1016/j.tins.2008.06.005
|
| [98] |
Schnurra I, Bernstein HG, Riederer P, et al. (2001) The neuronal calcium sensor protein VILIP-1 is associated with amyloid plaques and extracellular tangles in Alzheimer’s disease and promotes cell death and tau phosphorylation in vitro: a link between calcium sensors and Alzheimer's disease? Neurobiol Dis 8: 900-909. doi:10.1006/nbdi.2001.0432. doi: 10.1006/nbdi.2001.0432
|
| [99] |
Lee JM, Blennow K, Andreasen N, et al. (2008) The brain injury biomarker VLP-1 is increased in the cerebrospinal fluid of Alzheimer disease patients. Clin Chem 54: 1617-1623. doi:10.1373/clinchem.2008.104497. doi: 10.1373/clinchem.2008.104497
|
| [100] |
Liebl MP, Kaya AM, Tenzer S, et al. (2014) Dimerization of visinin-like protein 1 is regulated by oxidative stress and calcium and is a pathological hallmark of amyotrophic lateral sclerosis. Free Radic Biol Med 72: 41-54. doi:10.1016/j.freeradbiomed.2014.04.008. doi: 10.1016/j.freeradbiomed.2014.04.008
|
| [101] |
Stejskal D, Sporova L, Svestak M, et al. (2011) Determination of serum visinin like protein-1 and its potential for the diagnosis of brain injury due to the stroke: a pilot study. Biomed Pap Med Fac Univ Palacký Olomouc Czechoslov 155: 263-268. doi:10.5507/bp.2011.049. doi: 10.5507/bp.2011.049
|
| [102] |
Bernstein H-G, Braunewell K-H, Spilker C, et al. (2002) Hippocampal expression of the calcium sensor protein visinin-like protein-1 in schizophrenia. Neuroreport 13: 393-396. http://www.ncbi.nlm.nih.gov/pubmed/11930147. doi: 10.1097/00001756-200203250-00006
|
| [103] | Kester MI, Teunissen CE, Sutphen C, et al. Cerebrospinal fluid VILIP-1 and YKL-40, candidate biomarkers to diagnose, predict and monitor Alzheimer’s disease in a memory clinic cohort. Alzheimers Res Ther 7: 59. doi:10.1186/s13195-015-0142-1. |
| [104] |
Tarawneh R, D’Angelo G, Macy E, et al. (2011) Visinin-like protein-1: diagnostic and prognostic biomarker in Alzheimer disease. Ann Neurol 70: 274-285. doi:10.1002/ana.22448. doi: 10.1002/ana.22448
|
| [105] |
Lin Q, Cao Y, Gao J (2014) Serum calreticulin is a negative biomarker in patients with Alzheimer’s disease. Int J Mol Sci 15: 21740-21753. doi:10.3390/ijms151221740. doi: 10.3390/ijms151221740
|
| [106] | Wu J-C, Liang Z-Q, Qin Z-H (2006) Quality control system of the endoplasmic reticulum and related diseases. Acta Biochim Biophys Sin 38: 219-226. doi:10.1111/j.1745-7270.2006.00156.x. |
| [107] |
Bernard-Marissal N, Moumen a., Sunyach C, et al. (2012) Reduced Calreticulin Levels Link Endoplasmic Reticulum Stress and Fas-Triggered Cell Death in Motoneurons Vulnerable to ALS. J Neurosci 32: 4901-4912. doi:10.1523/JNEUROSCI.5431-11. doi: 10.1523/JNEUROSCI.5431-11.2012
|
| [108] | Gelebart P, Opas M, Michalak M (2004) Calreticulin, a Ca2+-binding chaperone of the endoplasmic reticulum. Int J Biochem Cell Biol 37: 260-266. doi:10.1016/j.biocel.2004.02.030. |
| [109] |
Peterson JR, Ora A, Van PN, et al. (1995) Transient, lectin-like association of calreticulin with folding intermediates of cellular and viral glycoproteins. Mol Biol Cell 6: 1173-1184. doi:10.1091/mbc.6.9.1173. doi: 10.1091/mbc.6.9.1173
|
| [110] |
Duus K, Hansen PR, Houen G (2008) Interaction of calreticulin with amyloid beta peptide 1–42. Protein Pept Lett 15: 103-107. http://www.ncbi.nlm.nih.gov/pubmed/18221019.. doi: 10.2174/092986608783330459
|
| [111] |
Erickson RR, Dunning LM, Olson DA, et al. (2005) In cerebrospinal fluid ER chaperones ERp57 and calreticulin bind beta-amyloid. Biochem Biophys Res Commun 332: 50-57. doi:10.1016/j.bbrc.2005.04.090. doi: 10.1016/j.bbrc.2005.04.090
|
| [112] |
Taguchi J, Fujii A, Fujino Y, et al. (2000) Different expression of calreticulin and immunoglobulin binding protein in Alzheimer’s disease brain. Acta Neuropathol 100: 153-160. doi:10.1007/s004019900165. doi: 10.1007/s004019900165
|
| [113] |
Luo X, Weber GA, Zheng J, et al. (2003) C1q-calreticulin induced oxidative neurotoxicity: Relevance for the neuropathogenesis of Alzheimer’s disease. J Neuroimmunol 135: 62-71. doi:10.1016/S0165-5728(02)00444-7. doi: 10.1016/S0165-5728(02)00444-7
|
| [114] |
Díez-Guerra FJ (2010) Neurogranin, a link between calcium/calmodulin and protein kinase C signaling in synaptic plasticity. IUBMB Life 62: 597-606. doi:10.1002/iub.357. doi: 10.1002/iub.357
|
| [115] |
Kester MI, Teunissen CE, Crimmins DL, et al. (2015) Neurogranin as a Cerebrospinal Fluid Biomarker for Synaptic Loss in Symptomatic Alzheimer Disease. JAMA Neurol 72: 1275-1280. doi:10.1001/jamaneurol.2015.1867. doi: 10.1001/jamaneurol.2015.1867
|
| [116] |
Fyfe I (2015) Alzheimer disease: neurogranin in the CSF signals early Alzheimer disease and predicts disease progression. Nat Rev Neurol 11: 609. doi:10.1038/nrneurol.2015.178. doi: 10.1038/nrneurol.2015.178
|
| [117] |
Portelius E, Zetterberg H, Skillbäck T, et al. (2015) Cerebrospinal fluid neurogranin: relation to cognition and neurodegeneration in Alzheimer’s disease. Brain 138: 3373-3385. doi:10.1093/brain/awv267. doi: 10.1093/brain/awv267
|
| [118] |
De Vos A, Jacobs D, Struyfs H, et al. (2015) C-terminal neurogranin is increased in cerebrospinal fluid but unchanged in plasma in Alzheimer’s disease. Alzheimers Dement 11: 1461-1469. doi:10.1016/j.jalz.2015.05.012. doi: 10.1016/j.jalz.2015.05.012
|
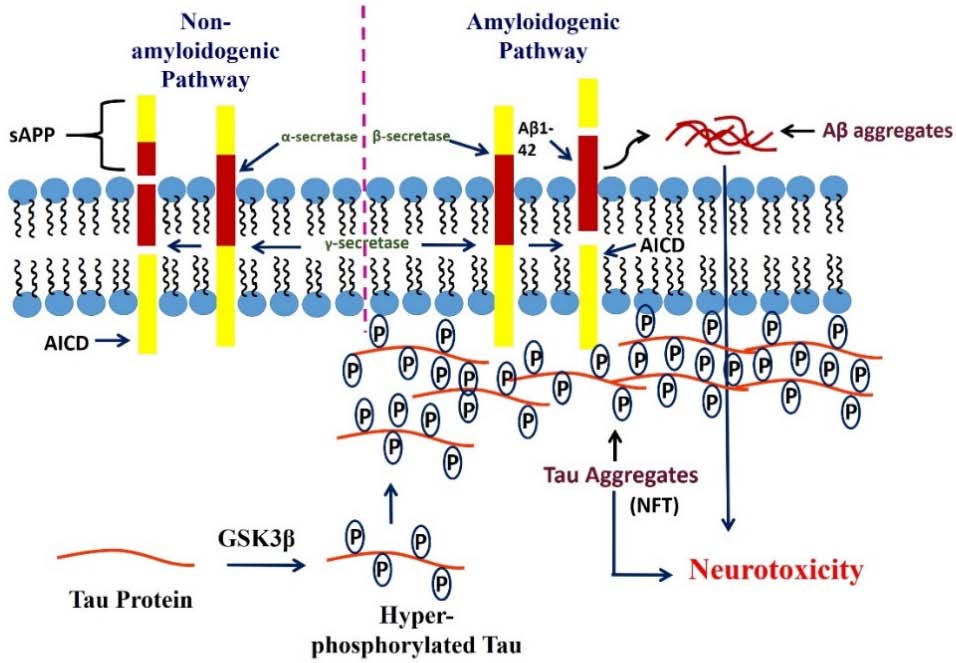
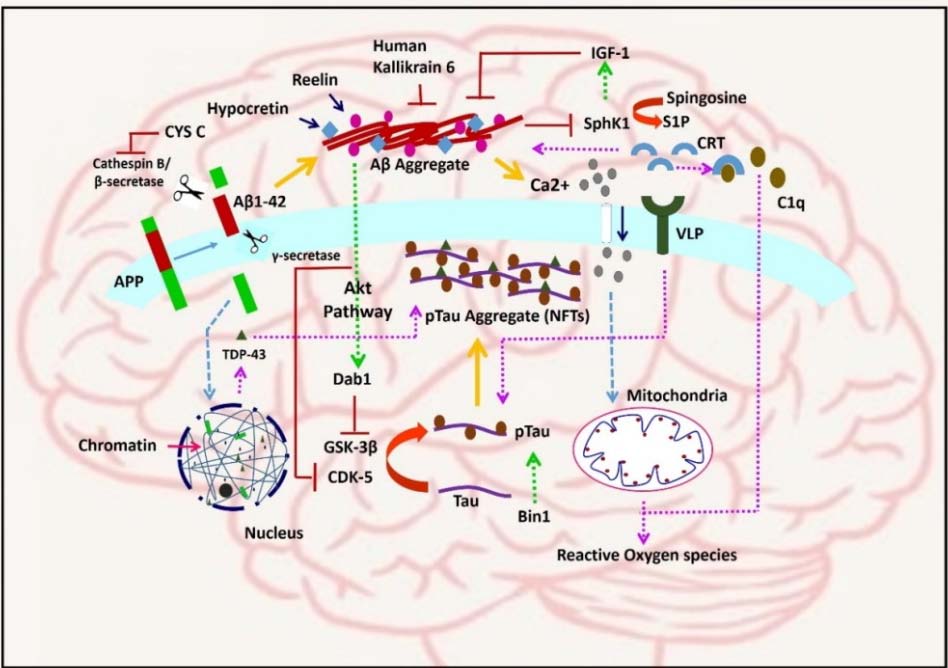
Figures(2)

Pravin D Potdar, Aashutosh U Shetti. Molecular Biomarkers for Diagnosis & Therapies of Alzheimer’s Disease[J]. AIMS Neuroscience, 2016, 3(4): 433-453. doi: 10.3934/Neuroscience.2016.4.433







 DownLoad:
DownLoad: