Citation: Lourdes A. Vega Rasgado, Arantxa Tabernero Urbieta, José María Medina Jiménez. Affected albumin endocytosis as a new neurotoxicity mechanism of amyloid beta[J]. AIMS Neuroscience, 2020, 7(3): 344-359. doi: 10.3934/Neuroscience.2020021

| [1] | Peterson BR (2008) Receptor-mediated endocytosis. The WileyEncyclopedia of Chemical Biology Hoboken, NJ: John Wiley and Sons Ltd, 1-13. |
| [2] |
Schmidt MR, Haucke V (2007) Recycling endosomes in neuronal membrane traffic. Biol Cell 99: 333-342. doi: 10.1042/BC20070007

|
| [3] |
Benmerah A, Lamaze C (2007) Clathrin-coated pits: vive la difference? Traffic 8: 970-982. doi: 10.1111/j.1600-0854.2007.00585.x
|
| [4] |
Sandvig K, Torgersen ML, Raa HA, et al. (2008) Clathrin-independent endocytosis: from nonexisting to an extreme degree of complexity. Histochem Cell Biol 129: 267-276. doi: 10.1007/s00418-007-0376-5
|
| [5] | Watson HA, Von Zastrow M, Wendland B (2004) Endocytosis. Encyclopedia of Molecular Cell Biology and Molecular Medicine Berlin, Germany: Wiley-VCH, 181-224. |
| [6] | Lentini D, Guzzi F, Pimpinelli F, et al. (2008) Polarization of caveolins and caveolae during migration of immortalized neurons. J Neurochem 104: 514-523. |
| [7] |
Nixon RA (2005) Endosome function and dysfunction in Alzheimer's disease and other neurodegenerative diseases. Neurobiol Aging 26: 373-382. doi: 10.1016/j.neurobiolaging.2004.09.018
|
| [8] |
Behl C, Davis JB, Lesley R, et al. (1994) Hydrogen peroxide mediates amyloid beta protein toxicity. Cell 77: 817-827. doi: 10.1016/0092-8674(94)90131-7
|
| [9] |
Harris ME, Hensley K, Butterfield DA, et al. (1995) Direct evidence of oxidative injury produced by the Alzheimer's beta-amyloid peptide (1–40) in cultured hippocampal neurons. Exp Neurol 131: 193-202. doi: 10.1016/0014-4886(95)90041-1
|
| [10] |
Bergin DH, Liu P (2010) Agmatine protects against beta-amyloid 25-35-induced memory impairments in the rat. Neuroscience 169: 794-811. doi: 10.1016/j.neuroscience.2010.05.004
|
| [11] |
Pike CJ, Walencewicz-Wasserman AJ, Kosmoski J, et al. (1995) Structure-activity analyses of beta-amyloid peptides: contributions of the beta 25-35 region to aggregation and neurotoxicity. J Neurochem 64: 253-265. doi: 10.1046/j.1471-4159.1995.64010253.x
|
| [12] |
Varadarajan S, Kanski J, Aksenova M, et al. (2001) Different mechanisms of oxidative stress and neurotoxicity for Alzheimer's A beta (1–42) and A beta (25−35). J Am Chem Soc 123: 5625-5631. doi: 10.1021/ja010452r
|
| [13] | Koudinov AR, Berezov TT (2004) Alzheimer's amyloid-beta (A beta) is an essential synaptic protein, not neurotoxic junk. Acta Neurobiol Exp (Wars) 64: 71-79. |
| [14] | Malyshev IY, Wiegant FA, Mashina SY, et al. (2005) Possible use of adaptation to hypoxia in Alzheimer's disease: a hypothesis. Med Sci Monit 11: HY31-HY38. |
| [15] |
Fishman PS, Farrand DA, Kristt DA (1990) Internalization of plasma proteins by cerebellar Purkinje cells. J Neurol Sci 100: 43-49. doi: 10.1016/0022-510X(90)90011-B
|
| [16] |
Granda B, Tabernero A, Tello V, et al. (2003) Oleic acid induces GAP-43 expression through a protein kinase C-mediated mechanism that is independent of NGF but synergistic with NT-3 and NT-4/5. Brain Res 988: 1-8. doi: 10.1016/S0006-8993(03)03253-0
|
| [17] |
Juurlink BH, Devon RM (1990) Macromolecular translocation—a possible function of astrocytes. Brain Res 533: 73-77. doi: 10.1016/0006-8993(90)91797-K
|
| [18] |
Tabernero A, Medina A, Sanchez-Abarca LI, et al. (1999) The effect of albumin on astrocyte energy metabolism is not brought about through the control of cytosolic Ca2+ concentrations but by free-fatty acid sequestration. Glia 25: 1-9. doi: 10.1002/(SICI)1098-1136(19990101)25:1<1::AID-GLIA1>3.0.CO;2-2
|
| [19] |
Vicario C, Medina JM (1992) Metabolism of lactate in the rat brain during the early neonatal period. J Neurochem 59: 32-40. doi: 10.1111/j.1471-4159.1992.tb08872.x
|
| [20] |
Tildon JT, McKenna MC, Stevenson J, et al. (1993) Transport of L-lactate by cultured rat brain astrocytes. Neurochem Res 18: 177-184. doi: 10.1007/BF01474682
|
| [21] |
Tabernero A, Granda B, Medina A, et al. (2002) Albumin promotes neuronal survival by increasing the synthesis and release of glutamate. J Neurochem 81: 881-891. doi: 10.1046/j.1471-4159.2002.00843.x
|
| [22] |
Puzzo D, Privitera L, Leznik E, et al. (2008) Picomolar amyloid-beta positively modulates synaptic plasticity and memory in hippocampus. J Neurosci 28: 14537-14545. doi: 10.1523/JNEUROSCI.2692-08.2008
|
| [23] |
Garcia-Osta A, Alberini CM (2009) Amyloid beta mediates memory formation. Learn Mem 16: 267-272. doi: 10.1101/lm.1310209
|
| [24] |
Morley JE, Farr SA, Banks WA, et al. (2010) A physiological role for amyloid-beta protein:enhancement of learning and memory. J Alzheimers Dis 19: 441-449. doi: 10.3233/JAD-2010-1230
|
| [25] |
Masters CL, Selkoe DJ (2012) Biochemistry of amyloid beta-protein and amyloid deposits in Alzheimer disease. Cold Spring Harbor Perspect Med 2: a006262. doi: 10.1101/cshperspect.a006262
|
| [26] |
Stevens RW, Elmendorf D, Gourlay M, et al. (1979) Application of fluoroimmunoassay to cerebrospinal fluid immunoglobulin G and albumin. J Clin Microbiol 10: 346-350. doi: 10.1128/JCM.10.3.346-350.1979
|
| [27] |
Daneman R, Prat A (2015) The blood-brain barrier. Cold Spring Harbor Perspect Biol 7: a020412. doi: 10.1101/cshperspect.a020412
|
| [28] |
Daneman R (2012) The blood-brain barrier in health and disease. Ann Neurol 72: 648-672. doi: 10.1002/ana.23648
|
| [29] |
Zlokovic BV (2008) The blood-brain barrier in health and chronic neurodegenerative disorders. Neuron 57: 178-201. doi: 10.1016/j.neuron.2008.01.003
|
| [30] |
Erickson MA, Banks WA (2013) Blood-brain barrier dysfunction as a cause and consequence of Alzheimer's disease. J Cereb Blood Flow Metab 33: 1500-1513. doi: 10.1038/jcbfm.2013.135
|
| [31] |
Zenaro E, Piacentino G, Constantin G (2017) The blood-brain barrier in Alzheimer's disease. Neurobiol Dis 107: 41-56. doi: 10.1016/j.nbd.2016.07.007
|
| [32] |
Zlokovic BV (2005) Neurovascular mechanisms of Alzheimer's neurodegeneration. Trends Neurosci 28: 202-208. doi: 10.1016/j.tins.2005.02.001
|
| [33] |
Bowman GL, Kaye JA, Moore M, et al. (2007) Blood-brain barrier impairment in Alzheimer disease: stability and functional significance. Neurology 68: 1809-1814. doi: 10.1212/01.wnl.0000262031.18018.1a
|
| [34] |
Vega L, Arroyo AA, Tabernero A, et al. (2009) Albumin-blunted deleterious effect of amyloid-beta by preventing the internalization of the peptide into neurons. J Alzheimers Dis 17: 795-805. doi: 10.3233/JAD-2009-1093
|
| [35] |
Shearman MS, Ragan CI, Iversen LL (1994) Inhibition of PC12 cell redox activity is a specific, early indicator of the mechanism of beta-amyloid-mediated cell death. Proc Natl Acad Sci USA 91: 1470-1474. doi: 10.1073/pnas.91.4.1470
|
| [36] | Megias L, Guerri C, Fornas E, et al. (2000) Endocytosis and transcytosis in growing astrocytes in primary culture. Possible implications in neural development. Int J Dev Biol 44: 209-221. |
| [37] |
Murk JL, Humbel BM, Ziese U, et al. (2003) Endosomal compartmentalization in three dimensions: implications for membrane fusion. Proc Natl Acad Sci USA 100: 13332-13337. doi: 10.1073/pnas.2232379100
|
| [38] |
Tabernero A, Bolanos JP, Medina JM (1993) Lipogenesis from lactate in rat neurons and astrocytes in primary culture. Biochem J 294: 635-638. doi: 10.1042/bj2940635
|
| [39] |
Denizot F, Lang R (1986) Rapid colorimetric assay for cell growth and survival. Modifications to the tetrazolium dye procedure giving improved sensitivity and reliability. J Immunol Methods 89: 271-277. doi: 10.1016/0022-1759(86)90368-6
|
| [40] |
Rosenkranz AR, Schmaldienst S, Stuhlmeier KM, et al. (1992) A microplate assay for the detection of oxidative products using 2′,7′-dichlorofluorescin-diacetate. J Immunol Methods 156: 39-45. doi: 10.1016/0022-1759(92)90008-H
|
| [41] |
Tabernero A, Lavado EM, Granda B, et al. (2001) Neuronal differentiation is triggered by oleic acid synthesized and released by astrocytes. J Neurochem 79: 606-616. doi: 10.1046/j.1471-4159.2001.00598.x
|
| [42] |
Lis H, Sela BA, Sachs L, et al. (1970) Specific inhibition by N-acetyl-D-galactosamine of the interaction between soybean agglutinin and animal cell surfaces. Biochim Biophys Acta 211: 582-585. doi: 10.1016/0005-2736(70)90265-8
|
| [43] |
Tabernero A, Velasco A, Granda B, et al. (2002) Transcytosis of albumin in astrocytes activates the sterol regulatory element-binding protein-1, which promotes the synthesis of the neurotrophic factor oleic acid. J Biol Chem 277: 4240-4246. doi: 10.1074/jbc.M108760200
|
| [44] |
Schnitzer JE, Carley WW, Palade GE (1988) Albumin interacts specifically with a 60-kDa microvascular endothelial glycoprotein. Proc Natl Acad Sci USA 85: 6773-6777. doi: 10.1073/pnas.85.18.6773
|
| [45] |
Crescenzi O, Tomaselli S, Guerrini R, et al. (2002) Solution structure of the Alzheimer amyloid beta-peptide (1–42) in an apolar microenvironment. Similarity with a virus fusion domain. Eur J Biochem 269: 5642-5648. doi: 10.1046/j.1432-1033.2002.03271.x
|
| [46] |
D'Ursi AM, Armenante MR, Guerrini R, et al. (2004) Solution structure of amyloid beta-peptide (25–35) in different media. J Med Chem 47: 4231-4238. doi: 10.1021/jm040773o
|
| [47] |
Chen GF, Xu TH, Yan Y, et al. (2017) Amyloid beta: structure, biology and structure-based therapeutic development. Acta Pharmacol Sin 38: 1205-1235. doi: 10.1038/aps.2017.28
|
| [48] |
Kaminsky YG, Marlatt MW, Smith MA, et al. (2010) Subcellular and metabolic examination of amyloid-beta peptides in Alzheimer disease pathogenesis: evidence for Abeta (25–35). Exp Neurol 221: 26-37. doi: 10.1016/j.expneurol.2009.09.005
|
| [49] | Pena F, Ordaz B, Balleza-Tapia H, et al. (2010) Beta-amyloid protein (25–35) disrupts hippocampal network activity: role of Fyn-kinase. Hippocampus 20: 78-96. |
| [50] |
Bi H, Sze CI (2002) N-methyl-D-aspartate receptor subunit NR2A and NR2B messenger RNA levels are altered in the hippocampus and entorhinal cortex in Alzheimer's disease. J Neurol Sci 200: 11-18. doi: 10.1016/S0022-510X(02)00087-4
|
| [51] |
Wesen E, Jeffries GDM, Matson Dzebo M, et al. (2017) Endocytic uptake of monomeric amyloid-beta peptides is clathrin- and dynamin-independent and results in selective accumulation of Abeta (1–42) compared to Abeta (1–40). Sci Rep 7: 2021. doi: 10.1038/s41598-017-02227-9
|
| [52] |
Glick JL (1991) Proposed mechanism for alteration of albumin structure and function in Alzheimer's disease. J Theor Biol 148: 283-286. doi: 10.1016/S0022-5193(05)80346-7
|
| [53] |
Liu Z, Liu J, Wang S, et al. (2016) Neuronal uptake of serum albumin is associated with neuron damage during the development of epilepsy. Exp Ther Med 12: 695-701. doi: 10.3892/etm.2016.3397
|
| [54] |
Hassel B, Iversen EG, Fonnum F (1994) Neurotoxicity of albumin in vivo. Neurosci Lett 167: 29-32. doi: 10.1016/0304-3940(94)91020-0
|
| [55] |
LeVine SM (2016) Albumin and multiple sclerosis. BMC Neurol 16: 47. doi: 10.1186/s12883-016-0564-9
|
| [56] |
Basi GS, Jacobson RD, Virag I, et al. (1987) Primary structure and transcriptional regulation of GAP-43, a protein associated with nerve growth. Cell 49: 785-791. doi: 10.1016/0092-8674(87)90616-7
|
| [57] |
Gorgels TG, Van Lookeren Campagne M, Oestreicher AB, et al. (1989) B-50/GAP43 is localized at the cytoplasmic side of the plasma membrane in developing and adult rat pyramidal tract. J Neurosci 9: 3861-3869. doi: 10.1523/JNEUROSCI.09-11-03861.1989
|
| [58] |
Huang SL, Merat D, Cheung WY (1989) Phosphatidylinositol modulates the response of calmodulin-dependent phosphatase to calmodulin. Arch Biochem Biophys 270: 42-49. doi: 10.1016/0003-9861(89)90005-2
|
| [59] | James G, Olson EN (1989) Identification of a novel fatty acylated protein that partitions between the plasma membrane and cytosol and is deacylated in response to serum and growth factor stimulation. J Biol Chem 264: 20998-21006. |
| [60] |
Jochen A, Hays J, Lianos E, et al. (1991) Insulin stimulates fatty acid acylation of adipocyte proteins. Biochem Biophys Res Commun 177: 797-801. doi: 10.1016/0006-291X(91)91859-B
|
| [61] |
Patterson SI, Skene JH (1999) A shift in protein S-palmitoylation, with persistence of growth-associated substrates, marks a critical period for synaptic plasticity in developing brain. J Neurobiol 39: 423-437. doi: 10.1002/(SICI)1097-4695(19990605)39:3<423::AID-NEU8>3.0.CO;2-Z
|
| [62] |
Rossi S, Furlan R, De Chiara V, et al. (2012) Interleukin-1 beta causes synaptic hyperexcitability in multiple sclerosis. Ann Neurol 71: 76-83. doi: 10.1002/ana.22512
|
| [63] |
Li V, Brustovetsky T, Brustovetsky N (2009) Role of cyclophilin D-dependent mitochondrial permeability transition in glutamate-induced calcium deregulation and excitotoxic neuronal death. Exp Neurol 218: 171-182. doi: 10.1016/j.expneurol.2009.02.007
|
| [64] |
Paul J, Strickland S, Melchor JP (2007) Fibrin deposition accelerates neurovascular damage and neuroinflammation in mouse models of Alzheimer's disease. J Exp Med 204: 1999-2008. doi: 10.1084/jem.20070304
|
| [65] |
Revett TJ, Baker GB, Jhamandas J, et al. (2013) Glutamate system, amyloid ss peptides and tau protein: functional interrelationships and relevance to Alzheimer disease pathology. J Psychiatry Neurosci 38: 6-23. doi: 10.1503/jpn.110190
|
| [66] |
Malik AR, Willnow TE (2019) Excitatory Amino Acid Transporters in Physiology and Disorders of the Central Nervous System. Int J Mol Sci 20: 5671. doi: 10.3390/ijms20225671
|
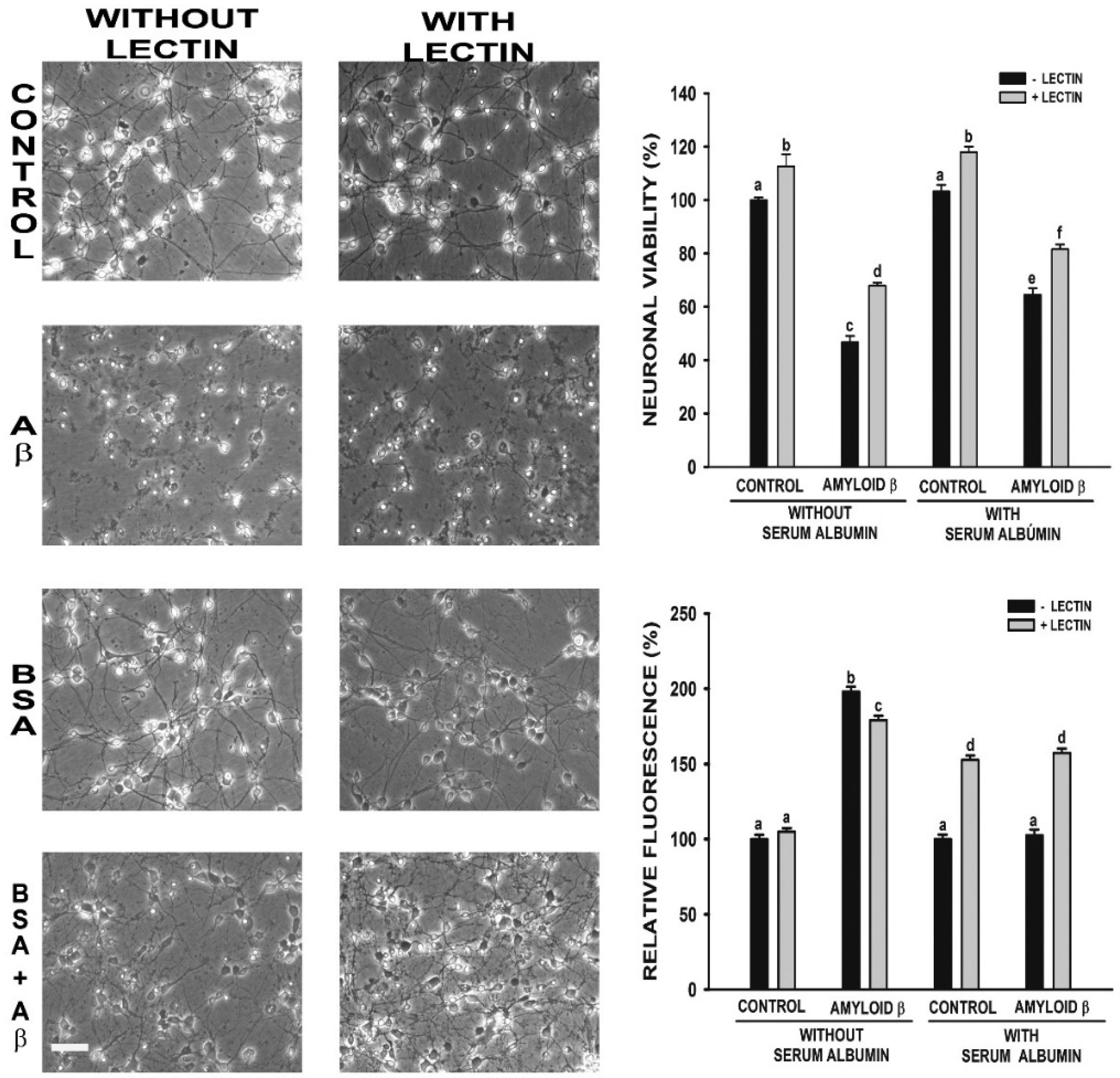
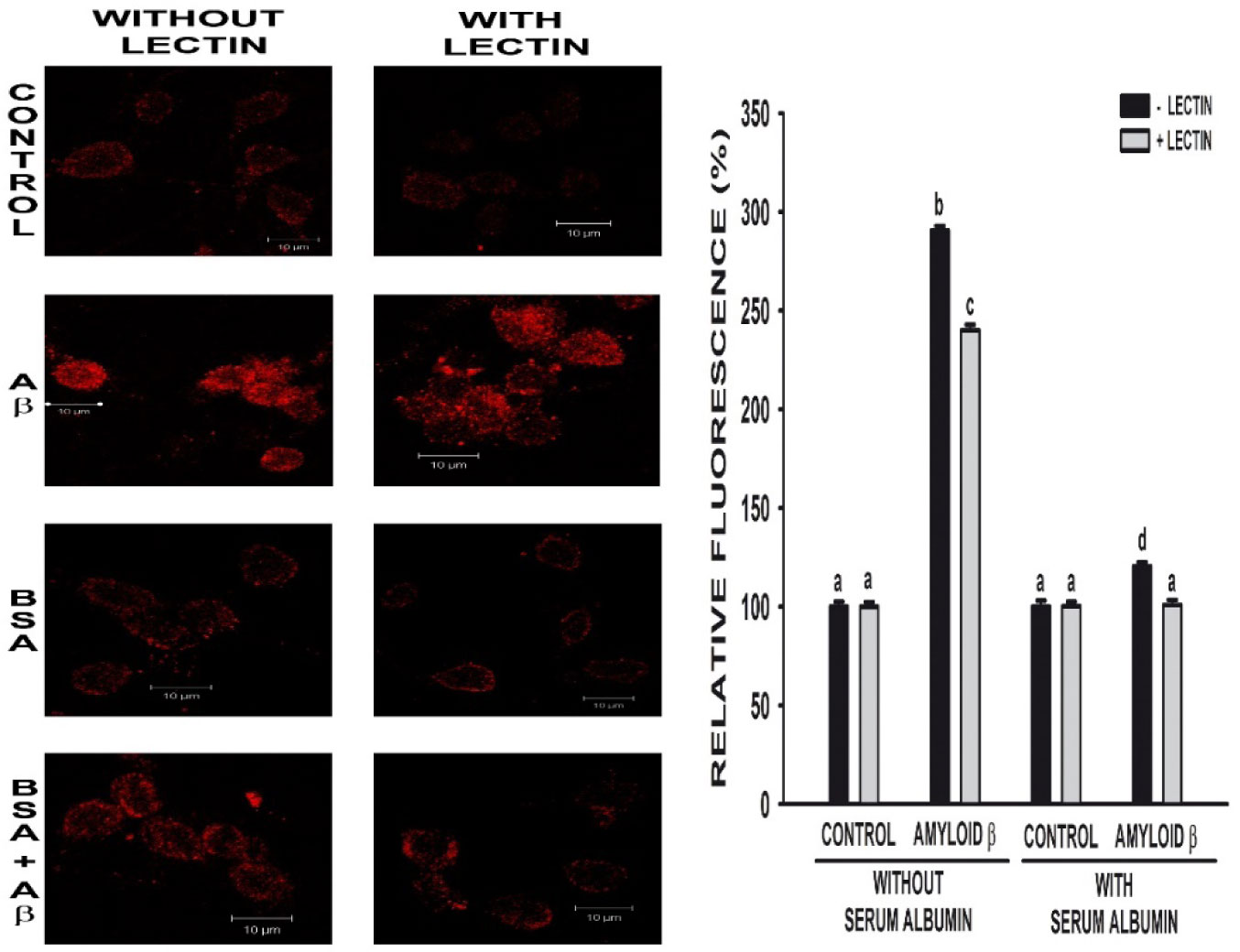
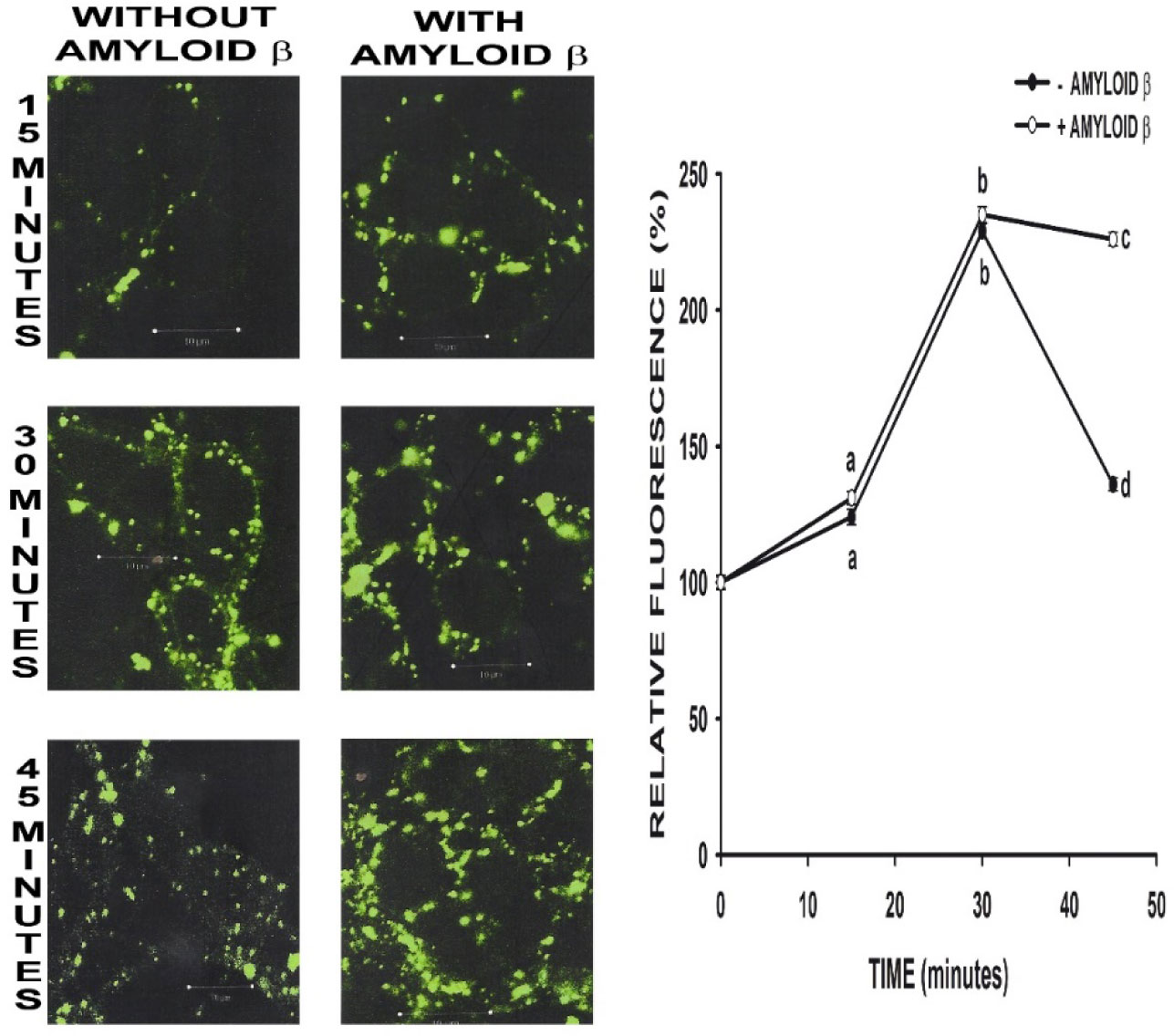
Figures(3)

Lourdes A. Vega Rasgado, Arantxa Tabernero Urbieta, José María Medina Jiménez. Affected albumin endocytosis as a new neurotoxicity mechanism of amyloid beta[J]. AIMS Neuroscience, 2020, 7(3): 344-359. doi: 10.3934/Neuroscience.2020021







 DownLoad:
DownLoad: