Epigenetics is the study of how cells control gene activity without changing the DNA sequence. Epigenetic changes affect how genes are turned on and off or expressed, and thus help regulate how cells in different parts of the body use the same genetic code. Errors in the epigenetic process can not only lead to abnormal gene activity or inactivity, but can also influence alternative splicing (AS) and could cause human diseases. Understanding of how epigenetic defects can affect human health, especially for neurological disorders, could suggest targets for therapeutic interventions. For such a purpose, the Lesch-Nyhan disease (LND) has been selected as a valuable model to study the genetic-epigenetic interplay, especially to explore the epistasis between the housekeeping hypoxanthine phosphoribosyltransferase 1 (HPRT1) and β-amyloid precursor protein (APP) genes. This review is structured as follows: we begin with an overview about the monogenetic neurological disorders associated with epigenetic changes; next, the current knowledge on HPRT1 and APP genes is provided; then, the epistasis between HPRT1 and APP genes related to the neurobehavioral syndrome in LND is described; and finally, we present the construction of expression vectors to study intermolecular interactions between the hypoxanthine-guanine phosphoribosyltransferase (HGprt) enzyme and APP in LND. Information obtained from such expression vectors would be useful for future directions to design therapies through epigenetic interventions.
Citation: Khue Vu Nguyen. Epigenetic modulation of human neurobiological disorders: Lesch-Nyhan disease as a model disorder[J]. AIMS Neuroscience, 2025, 12(2): 58-74. doi: 10.3934/Neuroscience.2025005
Epigenetics is the study of how cells control gene activity without changing the DNA sequence. Epigenetic changes affect how genes are turned on and off or expressed, and thus help regulate how cells in different parts of the body use the same genetic code. Errors in the epigenetic process can not only lead to abnormal gene activity or inactivity, but can also influence alternative splicing (AS) and could cause human diseases. Understanding of how epigenetic defects can affect human health, especially for neurological disorders, could suggest targets for therapeutic interventions. For such a purpose, the Lesch-Nyhan disease (LND) has been selected as a valuable model to study the genetic-epigenetic interplay, especially to explore the epistasis between the housekeeping hypoxanthine phosphoribosyltransferase 1 (HPRT1) and β-amyloid precursor protein (APP) genes. This review is structured as follows: we begin with an overview about the monogenetic neurological disorders associated with epigenetic changes; next, the current knowledge on HPRT1 and APP genes is provided; then, the epistasis between HPRT1 and APP genes related to the neurobehavioral syndrome in LND is described; and finally, we present the construction of expression vectors to study intermolecular interactions between the hypoxanthine-guanine phosphoribosyltransferase (HGprt) enzyme and APP in LND. Information obtained from such expression vectors would be useful for future directions to design therapies through epigenetic interventions.
Angiotensin-converting enzyme 2
acetyl coenzyme A
Alzheimer's disease
A disintegrin and metalloprotease-10
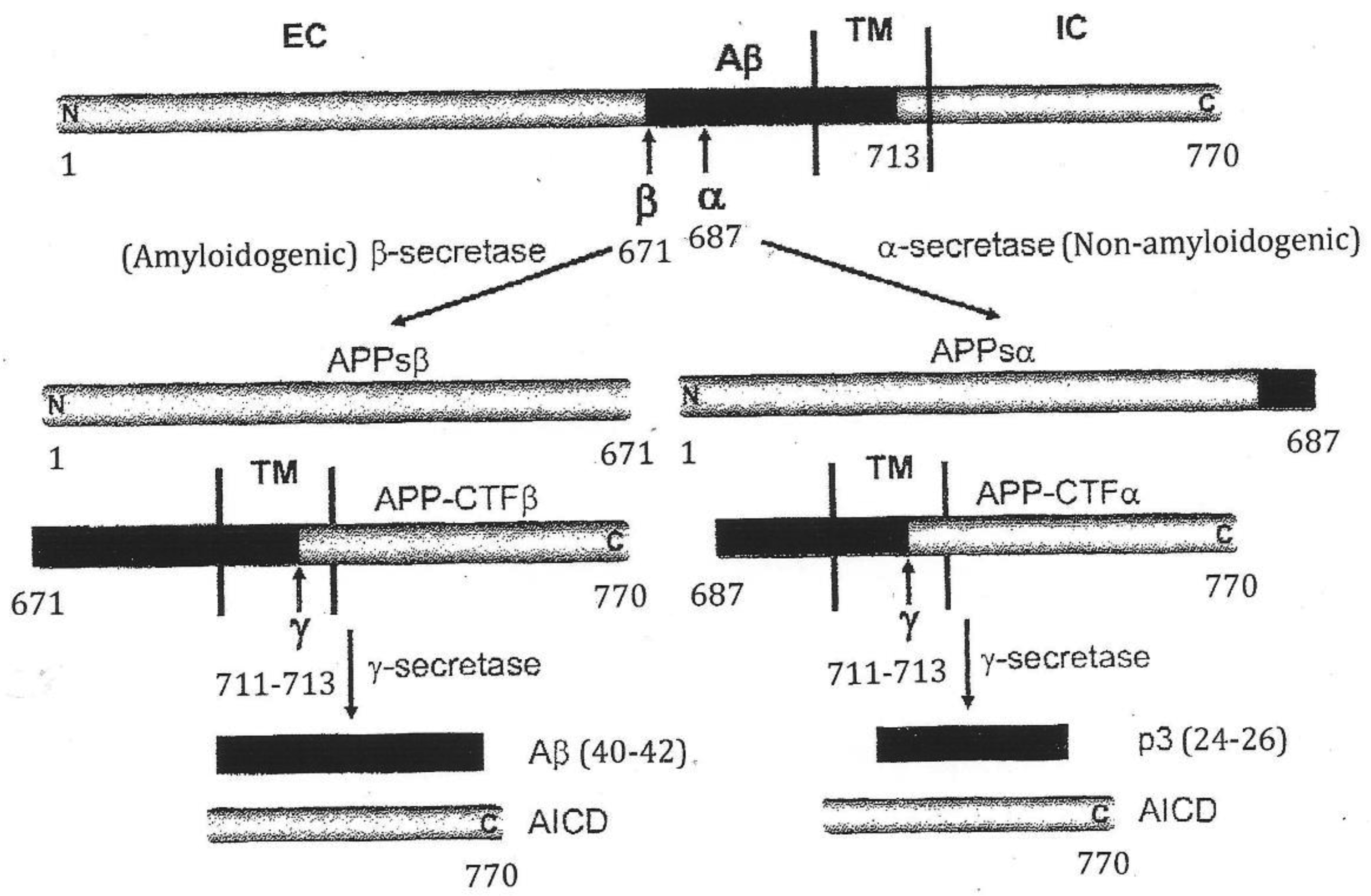
APP intracellular domain
Amyotrophic lateral sclerosis
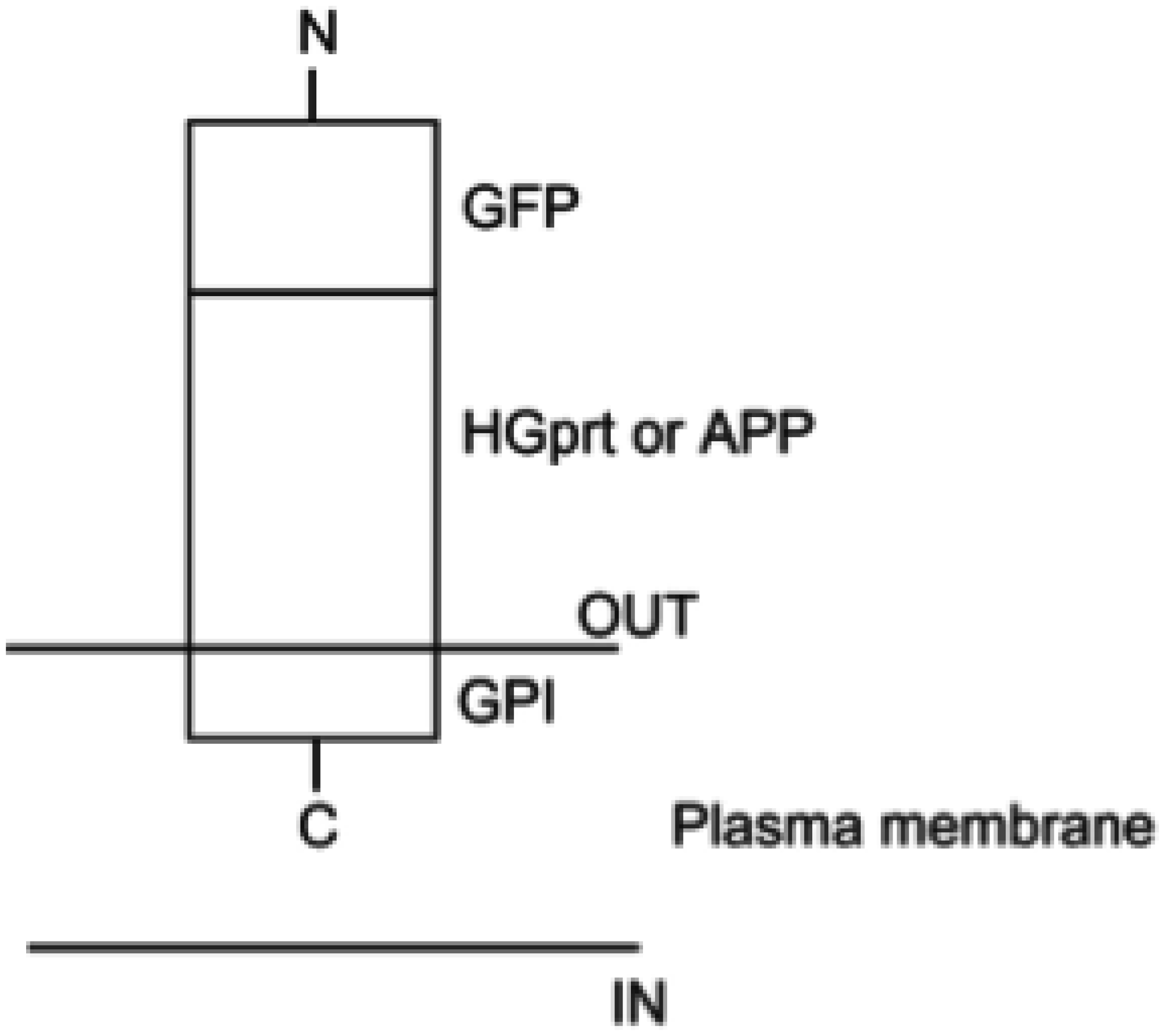
β-amyloid precursor protein
APP-like protein-2
Soluble APP derivative obtained after cleavage by α-secretase
Soluble APP derivative obtained after cleavage by β-secretase
APP-α-carboxyl-terminal fragment
APP-β-carboxyl-terminal fragment
Alternative splicing
Amyloid-β peptide
Alanine at position 713
cytidine analogues 5-azacytidine
β-site APP cleaving enzyme 1
Bromodomain and extraterminal
Carbonic anhydrase
entire coding sequence
Coronavirus disease 2019 virus
Central nervous system
Deoxyribonucleic acid
DNA methyltransferases
Extracellular domain
Euchromatic histone-lysine N-methyltransferase 1
Familial AD
human folate receptor 1
Fragile X syndrome
Green fluorescent protein
G9a-like protein
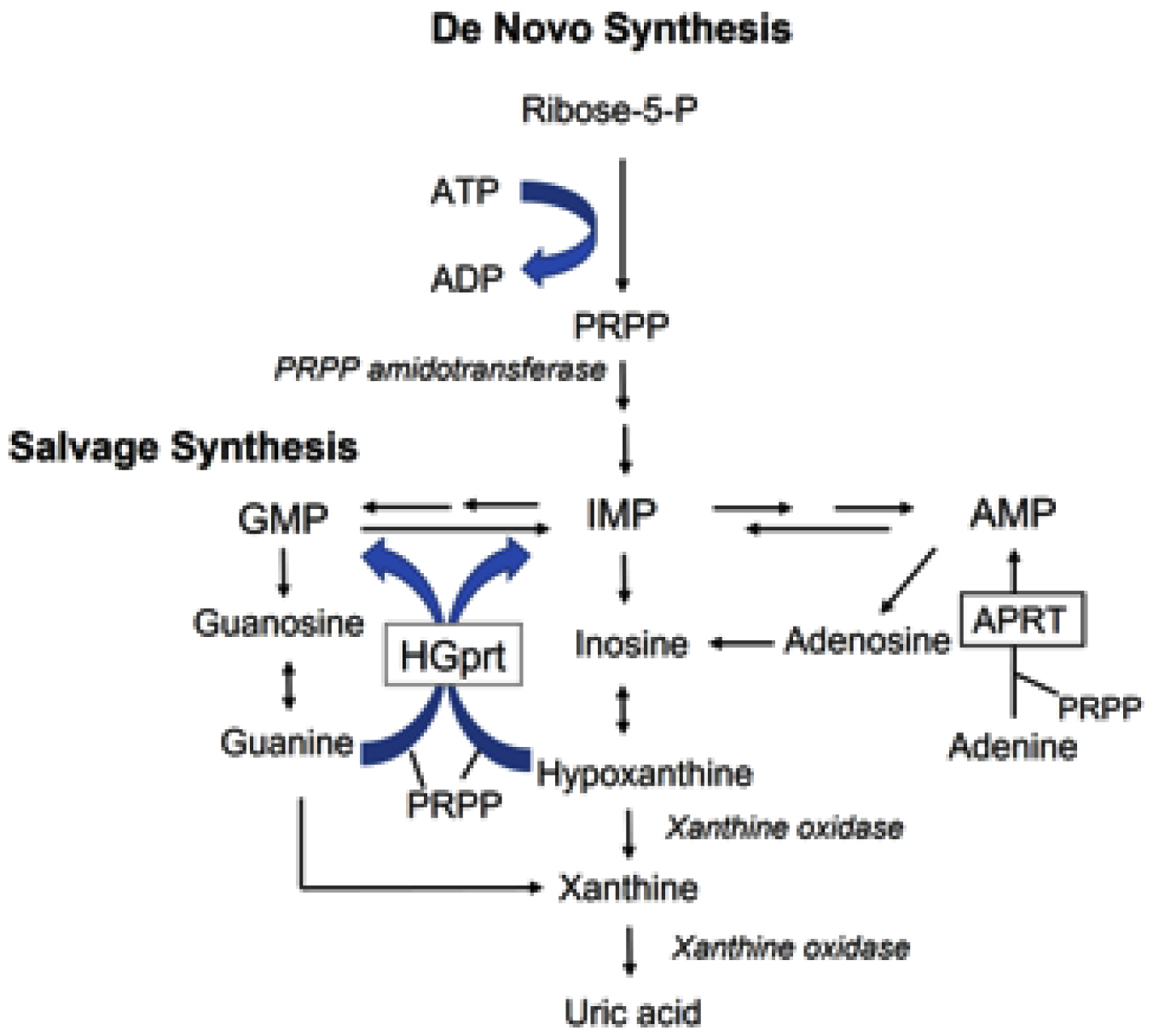
Guanosine monophosphate
Glycosylphosphatidylinositol
GPI-anchored proteins
Histone acetyltransferases
Histone deacetylases
Hypoxanthine-guanine phosphoribosyltransferase
Human leukocyte antigen
Hypoxanthine phosphoribosyltransferase 1
Histone methyltransferases
Intracellular domain
Inosine monophosphate
Deletion followed by an insertion
Kunitz-type serine protease inhibitor
Lysine at position 687
Lesch-Nyhan disease
Lessh-Nyhan variants
Methyl-CpG-binding
Methyl CpG binding protein 2
Mendelian inheritance in man
Messenger RNA
Multiple sclerosis
Methionine at position 671
Neurofibrillary tangles
National Institute of Neurological and Communicative Disorders and Stroke and the Alzheimer's disease and Related Disorder Association
phosphogluconate dehydrogenase
Precursor mRNA
Cellular prion protein (the ubiquitous normal cellular form)
α-D-5-phosphoribosyl-1-pyrophosphate
Presenilin
Ribonucleic acid
Sporadic AD
Severe acute respiratory syndrome coronavirus 2
Spike glycoprotein
Single-nucleotide polymorphism
Senile plaques
Transmembrane domain
Transmissible spongiform encephalopathies
Valine at position 711

| [1] |
Dupont C, Armant DR, Brenner CA (2009) Epigenetics: definition, mechanisms and clinical perspective. Semin Reprod Med 27: 351-357. https://doi.org/10.1055%2Fs-0029-1237423 
|
| [2] |
Mills JD, Nalpathamkalam T, Jacobs HI, et al. (2013) RNA-Seq analysis of the parietal cortex in Alzheimer's disease reveals alternatively spliced isoforms related to lipid metabolism. Neurosci Lett 536: 90-95. https://doi.org/10.1016/j.neulet.2012.12.042
|
| [3] |
Qian W, Liu F (2014) Regulation of alternative splicing of tau exon 10. Neurosci Bull 30: 367-377. https://doi.org/10.1007/s12264-013-1411-2
|
| [4] |
Jakovcevski M, Akbarian S (2012) Epigenetic mechanisms in neurodevelopmental and neurodegenerative disease. Nat Med 18: 1194-1204. https://doi.org/10.1038/nm.2828
|
| [5] |
Qureshi IA, Mehler MF (2013) Understanding neurological disease mechanisms in the era of epigenetics. JAMA Neurol 70: 703-710. https://doi.org/10.1001/jamaneurol.2013.1443
|
| [6] |
Christopher MA, Kyle SM, Katz DJ (2017) Neuroepigenetic mechanisms in disease. Epigenetics Chromatin 10: 47. https://doi.org/10.1186/s13072-017-0150-4
|
| [7] | Entrez Gene: MECP2 methyl CpG binding protein 2 (Rett syndrome). [cited 2025 April 09]. Available from: https://www.ncbi.nlm.nih.gov/gene?Db=gene&Cmd=ShowDetailView&TermToSearch=4204 |
| [8] |
Kleefstra T (2005) Disruption of the gene Euchromatin histone methyltransferase 1 (EuHMTase1) is associated with the 9q34 subtelomeric deletion syndrome. J Med Genet 42: 299-306. https://doi.org/10.1136/jmg.2004.028464
|
| [9] | Kleefstra T, de Leeuw N (2010) Kleefstra Syndrome. GeneReviews® [Internet] . Seattle (WA): University of Washington, Seattle 1993-2025. |
| [10] |
Ren R, Liu Y, Liu P, et al. (2024) Clinical characteristics and genetic analysis of four pediatric patients with Kleefstra syndrome. BMC Med Genomics 17: 290. https://doi.org/10.1186/s12920-024-02065-5
|
| [11] |
Suraweera A, O'Byrne KJ, Richard DJ (2025) Epigenetic drugs in cancer therapy. Cancer Metastasis Rev 44: 37. https://doi.org/10.1007/s10555-025-10253-7
|
| [12] |
Lesch M, Nyhan WL (1964) A familial disorder of uric acid metabolism and central nervous system function. Am J Med 36: 561-570. https://doi.org/10.1016/0002-9343(64)90104-4
|
| [13] |
Micheli V, Bertelli M, Jacomelli G, et al. (2018) Lesch-Nyhan disease: a rare disorder with many unresolved aspects. Medical University 1: 13-24. https://doi.org/10.2478/medu-2018-0002
|
| [14] |
Selkoe DJ, Podlisny MB, Joachim CL, et al. (1988) β-Amyloid precursor protein of Alzheimer disease occurs as 110-135-kilodalton membrane-associated proteins in neural and nonneural tissues. Proc Natl Acad Sci USA 85: 7341-7345. https://doi.org/10.1073/pnas.85.19.7341
|
| [15] |
Hampel H, Hardy J, Blennow K, et al. (2021) The amyloid-β pathway in Alzheimer's disease. Mol Psychiatry 26: 5481-5503. https://doi.org/10.1038/s41380-021-01249-0
|
| [16] |
Hong F, Yang S (2022) Amyloidosis in Alzheimer's disease: pathogeny, etiology, and related therapeutic directions. Molecules 27: 1210. https://doi.org/10.3390/molecules27041210
|
| [17] |
Orobets K, Karamyshev AL (2023) Amyloid precursor protein and Alzheimer's disease. Int J Mol Sci 24: 14794. https://doi.org/10.3390/ijms241914794
|
| [18] |
Zhang Y, Chen H, Li R, et al. (2023) Amyloid β-based therapy for Alzheimer's disease: challenges, successes and future. Signal Transduct Target Ther 8: 248. https://doi.org/10.1038/s41392-023-01484-7
|
| [19] |
Gottle M, Prudente CN, Fu R, et al. (2014) Loss of dopamine phenotype among midbrain neurons in Lesch-Nyhan disease. Ann Neurol 76: 95-107. https://doi.org/10.1002/ana.24191
|
| [20] |
Stacey NC, Ma MHY, Duley JA (2000) Abnormalities in cellular adhesion of neuroblastoma and fibroblast models of Lesch-Nyhan syndrome. Neuroscience 98: 397-401. https://doi.org/10.1016/S0306-4522(00)00149-4
|
| [21] |
Harris JC, Lee RR, Jinnah HA, et al. (1998) Craniocerebral magnetic resonance imaging measurement and findings in Lesch-Nyhan syndrome. Arch Neurol 55: 547-553. https://doi.org/10.1001/archneur.55.4.547
|
| [22] |
Schretlen DJ, Varvaris M, Ho TE, et al. (2013) Regional brain volume abnormalities in Lesch-Nyhan disease and its variants: a cross-sectional study. Lancet Neurol 12: 1151-1158. https://doi.org/10.1016/s1474-4422(13)70238-2
|
| [23] |
Schretlen DJ, Varvaris M, Vannorsdall TD, et al. (2015) Brain white matter volume abnormalities in Lesch-Nyhan disease and its variants. Neurology 84: 190-196. https://doi.org/10.1212/WNL.0000000000001128
|
| [24] |
Kang TH, Friedmann T (2015) Alzheimer's disease shares gene expression aberrations with purinergic dysregulation of HPRT deficiency (Lesch-Nyhan disease). Neurosci Lett 590: 35-39. https://doi.org/10.1016/j.neulet.2015.01.042
|
| [25] |
Nguyen KV (2014) Epigenetic regulation in amyloid precursor protein and the Lesch-Nyhan syndrome. Biochem Biophys Res Commun 446: 1091-1095. https://doi.org/10.1016/j.bbrc.2014.03.062
|
| [26] |
Nguyen KV (2015) Epigenetic regulation in amyloid precursor protein with genomic rearrangements and the Lesch-Nyhan syndrome. Nucleosides Nucleotides Nucleic Acids 34: 674-690. https://doi.org/10.1080/15257770.2015.1071844
|
| [27] |
Tuner PR, O'Connor K, Tate WP, et al. (2003) Roles of amyloid precursor protein and its fragments in regulating neural activity, plasticity and memory. Prog Neurobiol 70: 1-32. https://doi.org/10.1016/s0301-0082(03)00089-3
|
| [28] |
Pandey P, Sliker B, Peters HL, et al. (2016) Amyloid precursor protein and amycloid precursor-like protein 2 in cancer. Oncotarget 7: 19430-19444. https://doi.org/10.18632/oncotarget.7103
|
| [29] |
Zhao L, He D, Jiao M, et al. (2017) Overexpression of histone deacetylase and amyloid precursor protein in hepatocellular carcinoma. Technol Cancer Res Treat 16: 586-594. https://doi.org/10.1177/1533034616661664
|
| [30] | Gao L, Zhao H, Zhang D (2019) Role of APLP2 in the prognosis and clinicopathology of renal cell carcinoma. Oncol Lett 17: 508-513. https://doi.org/10.3892/ol.2018.9577 |
| [31] |
Imamura A, Yamanouchi H, Kurokawa T (1992) Elevated fibrinopeptide A (FPA) in patients with Lech-Nyhan syndrome. Brain Dev 14: 424-425. https://doi.org/10.1016/S0387-7604(12)80355-X
|
| [32] |
Riaz IB, Husnain M, Ateeli H (2014) Recurrent thrombosis in a patient with Lesch Nyhan syndrome. Am J Med 127: e11-e12. https://doi.org/10.1016/j.amjmed.2014.01.033
|
| [33] |
Tewari N, Mathur VP, Sardana D, et al. (2017) Lesch-Nyhan syndrome: the saga of metabolic abnormality and self-injurious behavior. Intractable Rare Dis Res 6: 65-68. https://doi.org/10.5582/irdr.2016.01076
|
| [34] |
Reisz JA, Dzieciatkowska M, Stephenson D, et al. (2023) Red blood cells from individuals with Lesch-Nyhan syndrome: multi-omics insights into a novel S162N mutation causing hypoxanthine-guanine phosphoribosyltransferase deficiency. Antioxidants 12: 1699. https://doi.org/10.3390/antiox12091699
|
| [35] |
Canobbio I, Visconte C, Momi S, et al. (2017) Platelet amyloid precursor protein is a modulator of venous thromboembolism in mice. Blood 130: 527-536. https://doi.org/10.1182/blood-2017-01-764910
|
| [36] |
Townsend MH, Felsted AM, Ence ZE, et al. (2017) Elevated expression of hypoxanthine guanine phosphoribosyltransferase within malignant tissue. Cancer Clin Oncol 6: 19. https://doi.org/10.5539/CCO.V6N2P19
|
| [37] |
Sedano MJ, Ramos EI, Choudhari R, et al. (2020) Hypoxanthine phosphoribosyltransferase 1 is upregulated, predicts clinical outcome and controls gene expression in breast cancer. Cancers 12: 1522. https://doi.org/10.3390/cancers12061522
|
| [38] |
Ahmadi M, Eftekhari Kenzerki M, Akrami SM, et al. (2021) Overexpression of HPRT1 is associated with poor prognosis in head and neck squamous cell carcinoma. FEBS Open Bio 11: 2525-2540. https://doi.org/10.1002/2211-5463.13250
|
| [39] |
Wu T, Jiao Z, Li Y, et al. (2022) HPRT1 promotes chemoresistance in oral squamous cell carcinoma via activating NMP1/PI3K/Akt signaling pathway. Cancers 14: 855. https://doi.org/10.3390/cancers14040855
|
| [40] |
Yuang L, Xiao Z, Lu R (2023) Hypoxanthine guanine phosphoribosyltransferase 1, a target of miR-125b-5p, promotes cell proliferation and invasion in head and neck squamous cell carcinoma. Heliyon 9: e20174. https://doi.org/10.1016/j.heliyon.2023.e20174
|
| [41] |
Chen A, Wang G, Wang D, et al. (2024) HPRT1: a preliminary investigation on its involvement in nasopharyngeal carcinoma. Discov Oncol 15: 624. https://doi.org/10.1007/s12672-024-01506-y
|
| [42] |
Weagel EG, Townsend MH, Anderson MD, et al. (2017) Unusual expression of HPRT on the surface of the colorectal cancer cell lines HT29 and SW620. Cancer Res 77: 2149. https://doi.org/10.1158/1538-7445.AM2017-2149
|
| [43] |
Townsend MH, Felsted AM, Burrup W, et al. (2018) Examination of hypoxanthine guanine phosphoribosyltransferase as a biomarker for colorectal cancer patients. Mol Cell Oncol 5: e1481810. https://doi.org/10.1080/23723556.2018.1481810
|
| [44] |
Townsend MH, Robison RA, O'Neill KL (2018) A review of HPRT and its emerging role in cancer. Med Oncol 35: 89. https://doi.org/10.1007/s12032-018-1144-1
|
| [45] |
Townsend MH, Shrestha G, Robison RA, et al. (2018) The expansion of targetable biomarkers for CAR T cell therapy. J Exp Clin Cancer Res 37: 163. https://doi.org/10.1186/s13046-018-0817-0
|
| [46] |
Townsend MH, Ence ZE, Felsted AM, et al. (2019) Potential new biomarkers for endometrial cancer. Cancer Cell Int 19: 19. https://doi.org/10.1186/s12935-019-0731-3
|
| [47] |
Townsend MH, Ence ZE, Cox TP, et al. (2020) Evaluation of the upregulation and surface expression of hypoxanthine guanine phosphoribosyltransferase in acute lymphoblastic leukemia and Burkitt's B cell lymphoma. Cancer Cell Int 20: 375. https://doi.org/10.1186/s12935-020-01457-8
|
| [48] |
Townsend MH, Bennion KB, Bitter E, et al. (2021) Overexpression and surface localization of HPRT in prostate cancer provides a potential target for cancer specific antibody mediated cellular cytotoxicity. Exp Cell Res 403: 112567. https://doi.org/10.1016/j.yexcr.2021.112567
|
| [49] |
Nguyen KV (2021) Potential molecular link between the β-amyloid precursor protein (APP) and hypoxanthine-guanine phosphoribosyltransferase (HGprt) enzyme in Lesch-Nyhan disease and cancer. AIMS Neurosci 8: 548-557. https://doi.org/10.3934/Neuroscience.2021030
|
| [50] |
Dawson PA, Gordon RB, Keough DT, et al. (2005) Normal HPRT coding region in a male with gout due to HPRT deficiency. Mol Genet Metab 85: 78-80. https://doi.org/10.1016/j.ymgme.2005.01.005
|
| [51] |
Garcia MG, Torres RJ, Prior C, et al. (2008) Normal HPRT coding region in complete and partial HPRT deficiency. Mol Genet Metab 94: 167-172. https://doi.org/10.1016/j.ymgme.2008.01.006
|
| [52] |
Nguyen KV, Naviaux RK, Paik KK, et al. (2012) Lesch-Nyhan syndrome: mRNA expression of HPRT in patients with enzyme proven deficiency of HPRT and normal HPRT coding region of DNA. Mol Genet Metab 106: 498-501. https://doi.org/10.1016/j.ymgme.2012.06.003
|
| [53] |
Fu R, Ceballos-Picot I, Torres RJ, et al. (2014) Genotype-phenotype correlations in neurogenetics: Lesch-Nyhan disease as a model disorder. Brain 147: 1282-1303. https://doi.org/10.1093/brain/awt202
|
| [54] |
Trigueros-Genau MT, Torres RJ (2014) From genotype to phenotype: clinical variability in Lesch-Nyhan disease and the role of epigenetics. Rev Clin Esp 214: 461-465. https://doi.org/j.rce.2014.03.018
|
| [55] |
Jinnah HA (2014) Lesch-nyhan disease with no HPRT1 gene mutation?. Rev Clin Esp 214: 459-460. https://doi.org/10.1016/j.rce.2014.04.007
|
| [56] | International Human Genome Sequencing Consortium.Finishing the euchromatic sequence of the human genome. Nature (2004) 431: 931-945. https://doi.org/10.1038/nature03001 |
| [57] |
Nguyen KV, Nyhan WL (2017) Quantification of various APP-mRNA isoforms and epistasis in Lesch-Nyhan disease. Neurosci Lett 643: 52-58. https://doi.org/10.1016/j.neulet.2017.02.016
|
| [58] |
Mukherjee S, Maddalena M, Lu Y, et al. (2022) Cross-talk between mutant p53 and p62/SQSTM1 augments cancer cell migration by promoting the degradation of cell adhesion proteins. Proc Natl Acad Sci USA 119: e2119644119. https://doi.org/10.1073/pnas.2119644119
|
| [59] |
Cordell HJ (2002) Epistasis: what it means, what it doesn't mean, and statistical method to detect it in humans. Hum Mol Genet 11: 2463-2468. https://doi.org/10.1093/hmg/11.20.2463
|
| [60] |
Moore JH (2003) The ubiquitous nature of epistasis in determining susceptibility to common human diseases. Hum Hered 56: 73-82. https://doi.org/10.1159/000073735
|
| [61] |
Moore JH (2005) A global view of epistasis. Nat Genet 37: 13-14. https://doi.org/10.1038/ng0105-13
|
| [62] |
Riordan JD, Nadeau JH (2017) From peas to disease: modifier genes, network resilience, and the genetics of health. Am J Hum Genet 101: 177-191. https://doi.org/10.1016/j.ajhg.2017.06.004
|
| [63] |
Sun H, Lan X, Ma L, et al. (2022) Revealing modifier variations characterizations for elucidating the genetic basis of human phenotypic variations. Hum Genet 141: 1223-1233. https://doi.org/10.1007/s00439-021-02362-4
|
| [64] |
Long Chau DD, Haang Ng LL, Zhai Y, et al. (2023) Amyloid precursor protein and its interacting proteins in neurodevelopment. Biochem Soc Trans 51: 1647-1659. https://doi.org/10.1042/BST20221527
|
| [65] |
Nguyen KV, Naviaux RK, Nyhan WL (2020) Lesch-Nyhan disease: I. Construction of expression vectors for hypoxanthine-guanine phosphoribosyltransferase (HGprt) enzyme and amyloid precursor protein (APP). Nucleosides Nucleotides Nucleic Acids 39: 905-922. https://doi.org/10.1080/15257770.2020.1714653
|
| [66] |
Yoshikai S, Sasaki H, Doh-ura K, et al. (1991) Genomic organization of the human human-amyloid β-protein precursor gene. Gene 102: 291-292. https://doi.org/10.1016/0378.1119(91)90093-q
|
| [67] |
Di Luca M, Colciaghi F, Pastorino L, et al. (2000) Platelets as a peripheral district where to study pathogenetic mechanisms of Alzheimer disease: the case of amyloid precursor protein. Eur J Pharmacol 405: 277-283. https://doi.org/10.1016/s0014-2999(00)00559-8
|
| [68] |
Ehehalt R, Keller P, Haass C, et al. (2003) Amyloidogenic processing of the Alzheimer β-amyloid precursor protein depends on lipid rafts. J Cell Biol 160: 113-123. https://doi.org/10.1083/jcb.200207113
|
| [69] |
Zheng H, Koo EH (2006) The amyloid precursor protein: beyond amyloid. Mol Neurodegener 1: 5. https://doi.org/10.1186/1750-1326-1-5
|
| [70] |
Connolly GP (1998) Fibroblasts models of neurological disorders: fluorescence measurement studies. Trends Pharmacol Sci 19: 171-177. https://doi.org/10.1016/s0165-6147(98)01202-4
|
| [71] |
Connor B, Firmin E, McCaughey-Chapman A, et al. (2018) Conversion of adult human fibroblasts into neural precursor cells using chemically modified mRNA. Heliyon 4: e00918. https://doi.org/10.1016/j.heliyon.2018.e00918
|
| [72] |
Yang Y, Chen R, Wu X, et al. (2019) Rapid efficient conversion of human fibroblasts into functional neurons by small molecules. Stem Cell Rep 13: 862-876. https://doi.org/10.1016/j.stemcr.2019.09.007
|
| [73] |
Hentschel A, Czech A, Munchberg U, et al. (2021) Protein signature of human skin fibroblasts allows the study of the molecular etiology of rare neurological diseases. Orphanet J Rare Dis 16: 73. https://doi.org/10.1186/s13023-020-01669-1
|
| [74] |
Linden R (2017) The biological function of the prion protein: a cell surface scaffold of signaling modules. Front Mol Neurosci 10: 77. https://doi.org/10.3389/fnmol.2017.00077
|
| [75] |
Lebreton S, Zurzolo C, Paladino S (2018) Organization of GPI-anchored proteins at the cell surface and its physiological relevance. Crit Rev Biochem Mol Biol 53: 403-419. https://doi.org/10.1080/10409238.2018.1485627
|
| [76] |
Germain P, Delalande A, Pichon C (2022) Role of muscle LIM protein in mechanotransduction process. Int J Mol Sci 23: 9785. https://doi.org/10.3390/ijms23179785
|
| [77] |
Choo SY (2007) The HLA system: genetics, immunology, clinical testing, and clinical implications. Yonsei Med J 48: 11-23. https://doi.org/10.3349/ymj.2007.48.1.11
|
| [78] |
Spiers L, Coupe N, Payne M (2019) Toxicities associated with checkpoint inhibitors - an overview. Rheumatology 58: vii7-vii16. https://doi.org/10.1093/rheumatology/kez418
|
| [79] |
Bauer AT, Gorzelanny C, Gebhardt C, et al. (2022) Interplay between coagulation and inflammation in cancer: limitations and therapeutic opportunities. Cancer Treat Rev 102: 102322. https://doi.org/10.1016/j.ctrv.2021.102322
|
| [80] |
Cantrell R, Palumbo JS (2022) Hemostasis and tumor immunity. Res Pract Thromb Haemost 6: e12728. https://doi.org/10.1002/rth2.12728
|
| [81] |
Coutard B, Valle C, de Lamballerie X, et al. (2020) The spike glycoprotein of the new coronavirus 2019-nCoV contains a furin-like cleavage site absent in CoV of the same clade. Antiviral Res 176: 104742. https://doi.org/10.1016/j.antiviral.2020.104742
|
| [82] |
Nguyen KV (2021) Problems associated with antiviral drugs and vaccine development for COVID-19: approach to intervention using expression vectors via GPI anchor. Nucleosides Nucleotides Nucleic Acids 40: 665-706. https://doi.org/10.1080/15257770.2021.1914851
|
| [83] |
Nguyen KV (2022) Containing the spread of COVID-19 virus facing to its high mutation rate: approach to intervention using a nonspecific way of blocking its entry into the cells. Nucleosides Nucleotides Nucleic Acids 41: 778-814. https://doi.org/10.1080/15257770.2022.2071937
|
| [84] |
Song YH, Kim CS, Seo JH (2016) Noninvasive monitoring of environmental toxicity through green fluorescent protein expressing Escherichia coli. Korea J Chem Eng 33: 1331-1336. https://doi.org/10.1007/s11814-015-0253-1
|
| [85] |
Talekar S, Jo BH, Dordick JS, et al. (2022) Carbonic anhydrase for CO2 capture, conversion and utilization. Curr Opin Biotechnol 74: 230-240. https://doi.org/10.1016/j.copbio.2021.12.003
|
| [86] |
Steger F, Reich J, Fuchs W, et al. (2022) Comparison of carbonic anhydrase for CO2 sequestration. Int J Mol Sci 23: 957. https://doi.org/10.3390/ijms23020957
|
| [87] |
Huili Z, Li K, Nguyen KV (2024) Climate change: approach to intervention using expression vector for carbonic anhydrase via glycosylphosphatidylinositol. Nucleosides Nucleotides Nucleic Acids 43: 134-145. https://doi.org/10.1080/15257770.2023.2238781
|
| [88] |
Lee D, Hong JH (2020) The fundamental role of bicarbonate transporters and associated carbonic anhydrase enzyme in maintaining ion and pH homeostasis in non-secretory organs. Int J Mol Sci 21: 339. https://doi.org/10.3390/ijms21010339
|
Figures(3)

Khue Vu Nguyen. Epigenetic modulation of human neurobiological disorders: Lesch-Nyhan disease as a model disorder[J]. AIMS Neuroscience, 2025, 12(2): 58-74. doi: 10.3934/Neuroscience.2025005







 DownLoad:
DownLoad: