Citation: Maria Kristina Parr, Anna Müller-Schöll. Pharmacology of doping agents—mechanisms promoting muscle hypertrophy[J]. AIMS Molecular Science, 2018, 5(2): 131-159. doi: 10.3934/molsci.2018.2.131

| [1] | World Anti-Doping Agency (2015) World anti-doping code. World Anti-Doping Agency. |
| [2] |
Shahidi NT (2001) A review of the chemistry, biological action, and clinical applications of anabolic-androgenic steroids. Clin Ther 23: 1355–1390. doi: 10.1016/S0149-2918(01)80114-4

|
| [3] |
Celotti F, Negri-Cesi P (1992) Anabolic-Steroids - a Review of Their Effects on the Muscles, of Their Possible Mechanisms of Action and of Their Use in Athletics. J Steroid Biochem Mol Biol 43: 469–477. doi: 10.1016/0960-0760(92)90085-W
|
| [4] | Dalbo VJ, Roberts MD, Mobley CB, et al. (2017) Testosterone and trenbolone enanthate increase mature myostatin protein expression despite increasing skeletal muscle hypertrophy and satellite cell number in rodent muscle. Andrologia 49. |
| [5] |
Bennett NC, Gardiner RA, Hooper JD, et al. (2010) Molecular cell biology of androgen receptor signalling. Int J Biochem Cell Biol 42: 813–827. doi: 10.1016/j.biocel.2009.11.013
|
| [6] |
Kicman AT (2008) Pharmacology of anabolic steroids. Br J Pharmacol 154: 502–521. doi: 10.1038/bjp.2008.165
|
| [7] |
Li J, Al-Azzawi F (2009) Mechanism of androgen receptor action. Maturitas 63: 142–148. doi: 10.1016/j.maturitas.2009.03.008
|
| [8] |
Maravelias C, Dona A, Stefanidou M, et al. (2005) Adverse effects of anabolic steroids in athletes. A constant threat. Toxicol Lett 158: 167–175. doi: 10.1016/j.toxlet.2005.06.005
|
| [9] |
Foradori CD, Weiser MJ, Handa RJ (2008) Non-genomic actions of androgens. Front Neuroendocrinol 29: 169–181. doi: 10.1016/j.yfrne.2007.10.005
|
| [10] | Kousteni S, Bellido T, Plotkin LI, et al. (2001) Nongenotropic, sex-nonspecific signaling through the estrogen or androgen receptors: Dissociation from transcriptional activity. Cell 104: 719–730. |
| [11] |
Hamdi MM, Mutungi G (2010) Dihydrotestosterone activates the MAPK pathway and modulates maximum isometric force through the EGF receptor in isolated intract mouse skeletal muscle fibres. J Physiol 588: 511–525. doi: 10.1113/jphysiol.2009.182162
|
| [12] |
Michels G, Hoppe UC (2008) Rapid actions of androgens. Front Neuroendocrinol 29: 182–198. doi: 10.1016/j.yfrne.2007.08.004
|
| [13] |
Norman AW, Mizwicki MT, Norman DP (2004) Steroid-hormone rapid actions, membrane receptors and a conformational ensemble model. Nat Rev Drug Discov 3: 27–41. doi: 10.1038/nrd1283
|
| [14] |
Rahman F, Christian HC (2007) Non-classical actions of testosterone: an update. Trends Endocrinol Metab 18: 371–378. doi: 10.1016/j.tem.2007.09.004
|
| [15] |
Losel RM, Falkenstein E, Feuring M, et al. (2003) Nongenomic steroid action: controversies, questions, and answers. Physiol Rev 83: 965–1016. doi: 10.1152/physrev.00003.2003
|
| [16] |
Mayer M, Rosen F (1977) Interaction of glucocorticoids and androgens with skeletal muscle. Metabolism 26: 937–962. doi: 10.1016/0026-0495(77)90013-0
|
| [17] |
Nicolaides NC, Galata Z, Kino T, et al. (2010) The human glucocorticoid receptor: molecular basis of biologic function. Steroids 75: 1–12. doi: 10.1016/j.steroids.2009.09.002
|
| [18] | Schaaf M, Cidlowski J (2003) Molecular mechanisms of glucocorticoid action and resistance. J Steroid Biochem Mol Biol 83: 37–48. |
| [19] |
Schoneveld OJ, Gaemers IC, Lamers WH (2004) Mechanisms of glucocorticoid signalling. Biochim Biophys Acta 1680: 114–128. doi: 10.1016/j.bbaexp.2004.09.004
|
| [20] |
Bhasin S, Storer TW, Berman N, et al. (1996) The effects of supraphysiologic doses of testosterone on muscle size and strength in normal men. N Engl J Med 335: 1–7. doi: 10.1056/NEJM199607043350101
|
| [21] |
Gorelick-Feldman J, Maclean D, Ilic N, et al. (2008) Phytoecdysteroids increase protein synthesis in skeletal muscle cells. J Agric Food Chem 56: 3532–3537. doi: 10.1021/jf073059z
|
| [22] |
van Amsterdam J, Opperhuizen A, Hartgens F (2010) Adverse health effects of anabolic-androgenic steroids. Regul Toxicol Pharmacol 57: 117–123. doi: 10.1016/j.yrtph.2010.02.001
|
| [23] | Mottram DR, George AJ (2000) Anabolic steroids. Baillieres Best Pract Res Clin Endocrinol Metab 14: 55–69. |
| [24] |
Achar S, Rostamian A, Narayan SM (2010) Cardiac and metabolic effects of anabolic-androgenic steroid abuse on lipids, blood pressure, left ventricular dimensions, and rhythm. Am J Cardiol 106: 893–901. doi: 10.1016/j.amjcard.2010.05.013
|
| [25] |
Herlitz LC, Markowitz GS, Farris AB, et al. (2010) Development of focal segmental glomerulosclerosis after anabolic steroid abuse. J Am Soc Nephrol 21: 163–172. doi: 10.1681/ASN.2009040450
|
| [26] |
Choi SM, Lee BM (2015) Comparative safety evaluation of selective androgen receptor modulators and anabolic androgenic steroids. Expert Opin Drug Saf 14: 1773–1785. doi: 10.1517/14740338.2015.1094052
|
| [27] |
Kicman AT, Gower DB (2003) Anabolic steroids in sport: biochemical, clinical and analytical perspectives. Ann Clin Biochem 40: 321–356. doi: 10.1258/000456303766476977
|
| [28] |
Basaria S (2010) Androgen abuse in athletes: detection and consequences. J Clin Endocrinol Metab 95: 1533–1543. doi: 10.1210/jc.2009-1579
|
| [29] |
Christou MA, Christou PA, Markozannes G, et al. (2017) Effects of Anabolic Androgenic Steroids on the Reproductive System of Athletes and Recreational Users: A Systematic Review and Meta-Analysis. Sports Med 47: 1869–1883. doi: 10.1007/s40279-017-0709-z
|
| [30] |
Rahnema CD, Lipshultz LI, Crosnoe LE, et al. (2014) Anabolic steroid-induced hypogonadism: diagnosis and treatment. Fertil Steril 101: 1271–1279. doi: 10.1016/j.fertnstert.2014.02.002
|
| [31] |
Nieschlag E, Vorona E (2015) Doping with anabolic androgenic steroids (AAS): Adverse effects on non-reproductive organs and functions. Rev Endocr Metab Disord 16: 199–211. doi: 10.1007/s11154-015-9320-5
|
| [32] |
Vasconsuelo A, Milanesi L, Boland R (2008) 17Beta-estradiol abrogates apoptosis in murine skeletal muscle cells through estrogen receptors: role of the phosphatidylinositol 3-kinase/Akt pathway. J Endocrinol 196: 385–397. doi: 10.1677/JOE-07-0250
|
| [33] |
Parr MK, Zhao P, Haupt O, et al. (2014) Estrogen receptor beta is involved in skeletal muscle hypertrophy induced by the phytoecdysteroid ecdysterone. Mol Nutr Food Res 58: 1861–1872. doi: 10.1002/mnfr.201300806
|
| [34] |
Weigt C, Hertrampf T, Zoth N, et al. (2012) Impact of estradiol, ER subtype specific agonists and genistein on energy homeostasis in a rat model of nutrition induced obesity. Mol Cell Endocrinol 351: 227–238. doi: 10.1016/j.mce.2011.12.013
|
| [35] |
Velloso CP (2008) Regulation of muscle mass by growth hormone and IGF-I. Br J Pharmacol 154: 557–568. doi: 10.1038/bjp.2008.153
|
| [36] |
Enmark E, Gustafsson JA (1999) Oestrogen receptors - an overview. J Intern Med 246: 133–138. doi: 10.1046/j.1365-2796.1999.00545.x
|
| [37] |
Weigt C, Hertrampf T, Kluxen FM, et al. (2013) Molecular effects of ER alpha- and beta-selective agonists on regulation of energy homeostasis in obese female Wistar rats. Mol Cell Endocrinol 377: 147–158. doi: 10.1016/j.mce.2013.07.007
|
| [38] |
Lazovic G, Radivojevic U, Milosevic V, et al. (2007) Tibolone and osteoporosis. Arch Gynecol Obstet 276: 577–581. doi: 10.1007/s00404-007-0387-4
|
| [39] |
Narayanan R, Coss CC, Dalton JT (2018) Development of selective androgen receptor modulators (SARMs). Mol Cell Endocrinol 465: 134–142. doi: 10.1016/j.mce.2017.06.013
|
| [40] |
Gao WQ, Dalton JT (2007) Expanding the therapeutic use of androgens via selective androgen receptor modulators (SARMs). Drug Discovery Today 12: 241–248. doi: 10.1016/j.drudis.2007.01.003
|
| [41] | Choo JJ, Horan MA, Little RA, et al. (1992) Anabolic effects of clenbuterol on skeletal muscle are mediated by beta 2-adrenoceptor activation. Am J Physiol 263: E50–56. |
| [42] | Moore N, Pegg G, Sillence M (1994) Anabolic effects of the beta2-andrenoceptor agonist salmeterol are dependent on route of administration. Am J Physiol 267: E475–E484. |
| [43] |
Barbosa J, Cruz C, Martins J, et al. (2005) Food poisoning by clenbuterol in Portugal. Food Addit Contam 22: 563–566. doi: 10.1080/02652030500135102
|
| [44] |
Brambilla G, Cenci T, Franconi F, et al. (2000) Clinical and pharmacological profile in a clenbuterol epidemic poisoning of contaminated beef meat in Italy. Toxicol Lett 114: 47–53. doi: 10.1016/S0378-4274(99)00270-2
|
| [45] | Sporano V, Grasso L, Esposito M, et al. (1998) Clenbuterol residues in non-liver containing meat as a cause of collective food poisoning. Vet Hum Toxicol 40: 141–143. |
| [46] | Brambilla G, Loizzo A, Fontana L, et al. (1997) Food poisoning following consumption of clenbuterol-treated veal in Italy. JAMA, J Am Med Assoc 278: 635. |
| [47] | Pulce C, Lamaison D, Keck G, et al. (1991) Collective human food poisonings by clenbuterol residues in veal liver. Vet Hum Toxicol 33: 480–481. |
| [48] | Salleras L, Dominguez A, Mata E, et al. (1995) Epidemiologic study of an outbreak of clenbuterol poisoning in Catalonia, Spain. Public Health Rep 110: 338–342. |
| [49] | Martinez-Navarro JF (1990) Food poisoning related to consumption of illicit beta-agonist in liver. Lancet 336: 1311. |
| [50] |
Parr MK, Blokland MH, Liebetrau F, et al. (2017) Distinction of clenbuterol intake from drug or contaminated food of animal origin in a controlled administration trial - the potential of enantiomeric separation for doping control analysis. Food Addit Contam Part A Chem Anal Control Expo Risk Assess 34: 525–535. doi: 10.1080/19440049.2016.1242169
|
| [51] |
Parr MK, Opfermann G, Schanzer W (2009) Analytical methods for the detection of clenbuterol. Bioanalysis 1: 437–450. doi: 10.4155/bio.09.29
|
| [52] |
Mersmann HJ (1998) Overview of the effects of beta-adrenergic receptor agonists on animal growth including mechanisms of action. J Anim Sci 76: 160–172. doi: 10.2527/1998.761160x
|
| [53] | World Anti-Doping Agency (2017) The 2018 Prohibited List. World Anti-Doping Agency. |
| [54] |
Serra C, Bhasin S, Tangherlini F, et al. (2011) The role of GH and IGF-I in mediating anabolic effects of testosterone on androgen-responsive muscle. Endocrinology 152: 193–206. doi: 10.1210/en.2010-0802
|
| [55] |
Rommel C, Bodine SC, Clarke BA, et al. (2001) Mediation of IGF-1-induced skeletal myotube hypertrophy by PI(3)K/Akt/mTOR and PI(3)K/Akt/GSK3 pathways. Nat Cell Biol 3: 1009–1013. doi: 10.1038/ncb1101-1009
|
| [56] |
Bolster DR, Jefferson LS, Kimball SR (2004) Regulation of protein synthesis associated with skeletal muscle hypertrophy by insulin-, amino acid- and exercise-induced signalling. Proc Nutr Soc 63: 351–356. doi: 10.1079/PNS2004355
|
| [57] |
Holecek M (2017) Beta-hydroxy-beta-methylbutyrate supplementation and skeletal muscle in healthy and muscle-wasting conditions. J Cachexia Sarcopenia Muscle 8: 529–541. doi: 10.1002/jcsm.12208
|
| [58] |
Thomas M, Langley B, Berry C, et al. (2000) Myostatin, a negative regulator of muscle growth, functions by inhibiting myoblast proliferation. J Biol Chem 275: 40235–40243. doi: 10.1074/jbc.M004356200
|
| [59] |
Schuelke M, Wagner KR, Stolz LE, et al. (2004) Myostatin mutation associated with gross muscle hypertrophy in a child. N Engl J Med 350: 2682–2688. doi: 10.1056/NEJMoa040933
|
| [60] |
McPherron AC, Lawler AM, Lee SJ (1997) Regulation of skeletal muscle mass in mice by a new TGF-beta superfamily member. Nature 387: 83–90. doi: 10.1038/387083a0
|
| [61] | Rı́os R, Carneiro I, Arce VM, et al. (2002) Myostatin is an inhibitor of myogenic differentiation. Am J Physiol Cell Physiol 282: C993–C999. |
| [62] |
Sharma M, Langley B, Bass J, et al. (2001) Myostatin in muscle growth and repair. Exerc Sport Sci Rev 29: 155–158. doi: 10.1097/00003677-200110000-00004
|
| [63] |
Joulia-Ekaza D, Cabello G (2007) The myostatin gene: physiology and pharmacological relevance. Curr Opin Pharmacol 7: 310–315. doi: 10.1016/j.coph.2006.11.011
|
| [64] |
Elliott B, Renshaw D, Getting S, et al. (2012) The central role of myostatin in skeletal muscle and whole body homeostasis. Acta Physiol 205: 324–340. doi: 10.1111/j.1748-1716.2012.02423.x
|
| [65] |
Gonzalez-Cadavid NF, Taylor WE, Yarasheski K, et al. (1998) Organization of the human myostatin gene and expression in healthy men and HIV-infected men with muscle wasting. Proc Natl Acad Sci USA 95: 14938–14943. doi: 10.1073/pnas.95.25.14938
|
| [66] |
Pearen MA, Ryall JG, Maxwell MA, et al. (2006) The orphan nuclear receptor, NOR-1, is a target of beta-adrenergic signaling in skeletal muscle. Endocrinology 147: 5217–5227. doi: 10.1210/en.2006-0447
|
| [67] |
Ma K, Mallidis C, Bhasin S, et al. (2003) Glucocorticoid-induced skeletal muscle atrophy is associated with upregulation of myostatin gene expression. Am J Physiol Endocrinol Metab 285: E363–E371. doi: 10.1152/ajpendo.00487.2002
|
| [68] |
Chen PR, Lee K (2016) INVITED REVIEW: Inhibitors of myostatin as methods of enhancing muscle growth and development. J Anim Sci 94: 3125–3134. doi: 10.2527/jas.2016-0532
|
| [69] |
Dschietzig TB (2014) Myostatin - From the Mighty Mouse to cardiovascular disease and cachexia. Clin Chim Acta 433: 216–224. doi: 10.1016/j.cca.2014.03.021
|
| [70] |
Kung T, Springer J, Doehner W, et al. (2010) Novel treatment approaches to cachexia and sarcopenia: highlights from the 5th Cachexia Conference. Expert Opin Investig Drugs 19: 579–585. doi: 10.1517/13543781003724690
|
| [71] |
Rodgers BD, Garikipati DK (2008) Clinical, agricultural, and evolutionary biology of myostatin: a comparative review. Endocr Rev 29: 513–534. doi: 10.1210/er.2008-0003
|
| [72] |
Marchitelli C, Savarese MC, Crisa A, et al. (2003) Double muscling in Marchigiana beef breed is caused by a stop codon in the third exon of myostatin gene. Mammalian Genome 14: 392–395. doi: 10.1007/s00335-002-2176-5
|
| [73] |
Bellinge RH, Liberles DA, Iaschi SP, et al. (2005) Myostatin and its implications on animal breeding: a review. Anim Genet 36: 1–6. doi: 10.1111/j.1365-2052.2004.01229.x
|
| [74] | Arnold H, Della-Fera M, Baile C (2001) Review of myostatin history, physiology and applications. Int Arch Biosci 1: 1014–1022. |
| [75] |
Lee YS, Lehar A, Sebald S, et al. (2015) Muscle hypertrophy induced by myostatin inhibition accelerates degeneration in dysferlinopathy. Hum Mol Genet 24: 5711–5719. doi: 10.1093/hmg/ddv288
|
| [76] | Bhasin S, Storer TW (2009) Anabolic applications of androgens for functional limitations associated with aging and chronic illness. Front Horm Res 37: 163–182. |
| [77] |
Dobs AS, Boccia RV, Croot CC, et al. (2013) Effects of enobosarm on muscle wasting and physical function in patients with cancer: a double-blind, randomised controlled phase 2 trial. Lancet Oncol 14: 335–345. doi: 10.1016/S1470-2045(13)70055-X
|
| [78] |
Dillon EL, Durham WJ, Urban RJ, et al. (2010) Hormone treatment and muscle anabolism during aging: androgens. Clin Nutr 29: 697–700. doi: 10.1016/j.clnu.2010.03.010
|
| [79] |
Madeddu C, Maccio A, Mantovani G (2012) Multitargeted treatment of cancer cachexia. Crit Rev Oncog 17: 305–314. doi: 10.1615/CritRevOncog.v17.i3.80
|
| [80] |
Ebner N, Springer J, Kalantar-Zadeh K, et al. (2013) Mechanism and novel therapeutic approaches to wasting in chronic disease. Maturitas 75: 199–206. doi: 10.1016/j.maturitas.2013.03.014
|
| [81] |
Jasuja R, LeBrasseur NK (2014) Regenerating skeletal muscle in the face of aging and disease. Am J Phys Med Rehabil 93: S88–96. doi: 10.1097/PHM.0000000000000118
|
| [82] |
Madeddu C, Mantovani G, Gramignano G, et al. (2015) Advances in pharmacologic strategies for cancer cachexia. Expert Opin Pharmacother 16: 2163–2177. doi: 10.1517/14656566.2015.1079621
|
| [83] |
Srinath R, Dobs A (2014) Enobosarm (GTx-024, S-22): a potential treatment for cachexia. Future Oncol 10: 187–194. doi: 10.2217/fon.13.273
|
| [84] |
Forbes JM, Cooper ME (2013) Mechanisms of diabetic complications. Physiol Rev 93: 137-188. doi: 10.1152/physrev.00045.2011
|
| [85] | Abdul-Ghani MA, DeFronzo RA (2010) Pathogenesis of insulin resistance in skeletal muscle. J Biomed Biotechnol 2010: 476279. |
| [86] |
Heinlein CA, Chang C (2002) Androgen receptor (AR) coregulatros: An overview. Endocr Rev 23: 175–200. doi: 10.1210/edrv.23.2.0460
|
| [87] |
Hengevoss J, Piechotta M, Muller D, et al. (2015) Combined effects of androgen anabolic steroids and physical activity on the hypothalamic-pituitary-gonadal axis. J Steroid Biochem Mol Biol 150: 86–96. doi: 10.1016/j.jsbmb.2015.03.003
|
| [88] | Murthy KS, Zhou H, Grider JR, et al. (2003) Inhibition of sustained smooth muscle contraction byPKA and PKG preferentially mediated by phosphorylation of RhoA. Am J Physiol Gastrointest Liver Physiol 284: G1006–1016. |
| [89] |
Mahabadi V, Amory JK, Swerdloff RS, et al. (2009) Combined transdermal testosterone gel and the progestin nestorone suppresses serum gonadotropins in men. J Clin Endocrinol Metab 94: 2313–2320. doi: 10.1210/jc.2008-2604
|
| [90] |
Bonsall RW, Rees HD, Michael RP (1985) The distribution, nuclear uptake and metabolism of [3H]dihydrotestosterone in the brain, pituitary gland and genital tract of the male rhesus monkey. J Steroid Biochem 23: 389–398. doi: 10.1016/0022-4731(85)90184-0
|
| [91] |
Bonsall RW, Rees HD, Michael RP (1983) Characterization by high performance liquid chromatography of nuclear metabolites of testosterone in the brains of male rhesus monkeys. Life Sci 33: 655–663. doi: 10.1016/0024-3205(83)90254-0
|
| [92] |
Tilbrook AJ, Clarke IJ (2001) Negative feedback regulation of the secretion and actions of gonadotropin-releasing hormone in males. Biol Reprod 64: 735–742. doi: 10.1095/biolreprod64.3.735
|
| [93] | Alen M, Reinila M, Vihko R (1985) Response of serum hormones to androgen administration in power athletes. Med Sci Sports Exerc 17: 354–359. |
| [94] |
Smith CL, O'Malley BW (2004) Coregulator function: a key to understanding tissue specificity of selective receptor modulators. Endocr Rev 25: 45–71. doi: 10.1210/er.2003-0023
|
| [95] |
McEwan IJ (2013) Androgen receptor modulators: a marriage of chemistry and biology. Future Med Chem 5: 1109–1120. doi: 10.4155/fmc.13.69
|
| [96] |
Narayanan R, Coss CC, Yepuru M, et al. (2008) Steroidal androgens and nonsteroidal, tissue-selective androgen receptor modulator, S-22, regulate androgen receptor function through distinct genomic and nongenomic signaling pathways. Mol Endocrinol 22: 2448–2465. doi: 10.1210/me.2008-0160
|
| [97] |
Skelton DA, Phillips SK, Bruce SA, et al. (1999) Hormone replacement therapy increases isometric muscle strength of adductor pollicis in post-menopausal women. Clin Sci 96: 357–364. doi: 10.1042/cs0960357
|
| [98] | Heikkinen J, Kyllonen E, Kurttila-Matero E, et al. (1997) HRT and exercise: effects on bone density, muscle strength and lipid metabolism. A placebo controlled 2-year prospective trial on two estrogen-progestin regimens in healthy postmenopausal women. Maturitas 26: 139–149. |
| [99] |
Glenmark B, Nilsson M, Gao H, et al. (2004) Difference in skeletal muscle function in males vs. females: role of estrogen receptor-beta. Am J Physiol Endocrinol Metab 287: E1125–1131. doi: 10.1152/ajpendo.00098.2004
|
| [100] |
Velders M, Schleipen B, Fritzemeier KH, et al. (2012) Selective estrogen receptor-beta activation stimulates skeletal muscle growth and regeneration. FASEB J 26: 1909–1920. doi: 10.1096/fj.11-194779
|
| [101] |
Harris HA, Albert LM, Leathurby Y, et al. (2003) Evaluation of an estrogen receptor-beta agonist in animal models of human disease. Endocrinology 144: 4241–4249. doi: 10.1210/en.2003-0550
|
| [102] |
Martin C, Ross M, Chapman KE, et al. (2004) CYP7B generates a selective estrogen receptor beta agonist in human prostate. J Clin Endocrinol Metab 89: 2928–2935. doi: 10.1210/jc.2003-031847
|
| [103] |
Harris HA, Bruner-Tran KL, Zhang X, et al. (2005) A selective estrogen receptor-beta agonist causes lesion regression in an experimentally induced model of endometriosis. Hum Reprod 20: 936–941. doi: 10.1093/humrep/deh711
|
| [104] |
Norman BH, Dodge JA, Richardson TI, et al. (2006) Benzopyrans are selective estrogen receptor beta agonists with novel activity in models of benign prostatic hyperplasia. J Med Chem 49: 6155–6157. doi: 10.1021/jm060491j
|
| [105] |
Mersereau JE, Levy N, Staub RE, et al. (2008) Liquiritigenin is a plant-derived highly selective estrogen receptor beta agonist. Mol Cell Endocrinol 283: 49–57. doi: 10.1016/j.mce.2007.11.020
|
| [106] |
Piu F, Cheevers C, Hyldtoft L, et al. (2008) Broad modulation of neuropathic pain states by a selective estrogen receptor beta agonist. Eur J Pharmacol 590: 423–429. doi: 10.1016/j.ejphar.2008.05.015
|
| [107] | Stovall DW, Pinkerton JV (2009) MF-101, an estrogen receptor beta agonist for the treatment of vasomotor symptoms in peri- and postmenopausal women. Curr Opin Investig Drugs 10: 365–371. |
| [108] |
Roman-Blas JA, Castaneda S, Cutolo M, et al. (2010) Efficacy and safety of a selective estrogen receptor beta agonist, ERB-041, in patients with rheumatoid arthritis: a 12-week, randomized, placebo-controlled, phase II study. Arthritis Care Res 62: 1588–1593. doi: 10.1002/acr.20275
|
| [109] |
Robb EL, Stuart JA (2011) Resveratrol interacts with estrogen receptor-beta to inhibit cell replicative growth and enhance stress resistance by upregulating mitochondrial superoxide dismutase. Free Radic Biol Med 50: 821–831. doi: 10.1016/j.freeradbiomed.2010.12.038
|
| [110] | Tagliaferri MA, Tagliaferri MC, Creasman JM, et al. (2016) A Selective Estrogen Receptor Beta Agonist for the Treatment of Hot Flushes: Phase 2 Clinical Trial. J Altern Complement Med 22: 722–728. |
| [111] |
Leitman DC, Christians U (2012) MF101: a multi-component botanical selective estrogen receptor beta modulator for the treatment of menopausal vasomotor symptoms. Expert Opin Investig Drugs 21: 1031–1042. doi: 10.1517/13543784.2012.685652
|
| [112] |
Jackson RL, Greiwe JS, Schwen RJ (2011) Emerging evidence of the health benefits of S-equol, an estrogen receptor beta agonist. Nutr Rev 69: 432–448. doi: 10.1111/j.1753-4887.2011.00400.x
|
| [113] |
Weiser MJ, Wu TJ, Handa RJ (2009) Estrogen receptor-beta agonist diarylpropionitrile: biological activities of R- and S-enantiomers on behavior and hormonal response to stress. Endocrinology 150: 1817–1825. doi: 10.1210/en.2008-1355
|
| [114] |
Amer DA, Kretzschmar G, Muller N, et al. (2010) Activation of transgenic estrogen receptor-beta by selected phytoestrogens in a stably transduced rat serotonergic cell line. J Steroid Biochem Mol Biol 120: 208–217. doi: 10.1016/j.jsbmb.2010.04.018
|
| [115] | Moutsatsou P (2007) The spectrum of phytoestrogens in nature: our knowledge is expanding. Hormones 6: 173–193. |
| [116] |
Arias-Loza PA, Hu K, Dienesch C, et al. (2007) Both estrogen receptor subtypes, alpha and beta, attenuate cardiovascular remodeling in aldosterone salt-treated rats. Hypertension 50: 432–438. doi: 10.1161/HYPERTENSIONAHA.106.084798
|
| [117] |
Sun J, Baudry J, Katzenellenbogen JA, et al. (2003) Molecular basis for the subtype discrimination of the estrogen receptor-beta-selective ligand, diarylpropionitrile. Mol Endocrinol 17: 247–258. doi: 10.1210/me.2002-0341
|
| [118] |
Escande A, Pillon A, Servant N, et al. (2006) Evaluation of ligand selectivity using reporter cell lines stably expressing estrogen receptor alpha or beta. Biochem Pharmacol 71: 1459–1469. doi: 10.1016/j.bcp.2006.02.002
|
| [119] |
Ogawa M, Kitano T, Kawata N, et al. (2017) Daidzein down-regulates ubiquitin-specific protease 19 expression through estrogen receptor beta and increases skeletal muscle mass in young female mice. J Nutr Biochem 49: 63–70. doi: 10.1016/j.jnutbio.2017.07.017
|
| [120] |
Choi SY, Ha TY, Ahn JY, et al. (2008) Estrogenic activities of isoflavones and flavones and their structure-activity relationships. Planta Med 74: 25–32. doi: 10.1055/s-2007-993760
|
| [121] |
Zierau O, Kolba S, Olff S, et al. (2006) Analysis of the promoter-specific estrogenic potency of the phytoestrogens genistein, daidzein and coumestrol. Planta Med 72: 184–186. doi: 10.1055/s-2005-873182
|
| [122] | Parr MK, Botre F, Nass A, et al. (2015) Ecdysteroids: A novel class of anabolic agents? Biol Sport 32: 169–173. |
| [123] |
Kohara A, Machida M, Setoguchi Y, et al. (2017) Enzymatically modified isoquercitrin supplementation intensifies plantaris muscle fiber hypertrophy in functionally overloaded mice. J Int Soc Sports Nutr 14: 32. doi: 10.1186/s12970-017-0190-y
|
| [124] |
Foryst-Ludwig A, Clemenz M, Hohmann S, et al. (2008) Metabolic actions of estrogen receptor beta (ERbeta) are mediated by a negative cross-talk with PPARgamma. PLoS Genet 4: e1000108. doi: 10.1371/journal.pgen.1000108
|
| [125] |
Semple RK, Chatterjee VK, O'Rahilly S (2006) PPAR gamma and human metabolic disease. J Clin Invest 116: 581–589. doi: 10.1172/JCI28003
|
| [126] |
Glass DJ (2005) Skeletal muscle hypertrophy and atrophy signaling pathways. Int J Biochem Cell Biol 37: 1974–1984. doi: 10.1016/j.biocel.2005.04.018
|
| [127] |
Bodine SC, Stitt TN, Gonzalez M, et al. (2001) Akt/mTOR pathway is a crucial regulator of skeletal muscle hypertrophy and can prevent muscle atrophy in vivo. Nat Cell Biol 3: 1014–1019. doi: 10.1038/ncb1101-1014
|
| [128] |
Wang M, Wang Y, Weil B, et al. (2009) Estrogen receptor beta mediates increased activation of PI3K/Akt signaling and improved myocardial function in female hearts following acute ischemia. Am J Physiol Regul Integr Comp Physiol 296: R972–978. doi: 10.1152/ajpregu.00045.2009
|
| [129] |
Stitt TN, Drujan D, Clarke BA, et al. (2004) The IGF-1/PI3K/Akt pathway prevents expression of muscle atrophy-induced ubiquitin ligases by inhibiting FOXO transcription factors. Mol Cell 14: 395–403. doi: 10.1016/S1097-2765(04)00211-4
|
| [130] |
Guttdrige D (2004) Signaling pathways weigh in on decisions to make or break skeletal muscle. Curr Opin Clin Nutr Metab Care 7: 443–450. doi: 10.1097/01.mco.0000134364.61406.26
|
| [131] |
Nader GA (2005) Molecular determinants of skeletal muscle mass: getting the "AKT" together. Int J Biochem Cell Biol 37: 1985–1996. doi: 10.1016/j.biocel.2005.02.026
|
| [132] |
Glass DJ (2010) Signaling pathways perturbing muscle mass. Curr Opin Clin Nutr Metab Care 13: 225–229. doi: 10.1097/MCO.0b013e32833862df
|
| [133] |
Song YH, Godard M, Li Y, et al. (2005) Insulin-like growth factor I-mediated skeletal muscle hypertrophy is characterized by increased mTOR-p70S6K signaling without increased Akt phosphorylation. J Investig Med 53: 135–142. doi: 10.2310/6650.2005.00309
|
| [134] |
Glass DJ (2003) Signalling pathways that mediate skeletal muscle hypertrophy and atrophy. Nat Cell Biol 5: 87–90. doi: 10.1038/ncb0203-87
|
| [135] |
Schiaffino S, Mammucari C (2011) Regulation of skeletal muscle growth by the IGF1-Akt/PKB pathway: insights from genetic models. Skelet Muscle 1: 4. doi: 10.1186/2044-5040-1-4
|
| [136] |
Lux MP, Fasching PA, Schrauder MG, et al. (2016) The PI3K Pathway: Background and Treatment Approaches. Breast Care 11: 398–404. doi: 10.1159/000453133
|
| [137] |
MacLennan P, Edwards R (1989) Effects of clenbuterol and propanolol on muscle mass. Biochem J 264: 573–579. doi: 10.1042/bj2640573
|
| [138] | Schmidt P, Holsboer F, Spengler D (2001) β2-Adrenergic Receptors Potentiate Glucocorticoid Receptor Transactivation via G Proteinβγ-Subunits and the Phosphoinositide 3-Kinase Pathway. Mol Endocrinol 15: 553–564. |
| [139] |
Murga C, Laguinge L, Wetzker R, et al. (1998) Activation of Akt/Protein Kinase B by G Protein-coupled Receptors A ROLE FOR α AND βγ SUBUNITS OF HETEROTRIMERIC G PROTEINS ACTING THROUGH PHOSPHATIDYLINOSITOL-3-OH KINASEγ. J Biol Chem 273: 19080–19085. doi: 10.1074/jbc.273.30.19080
|
| [140] | Huang P, Li Y, Lv Z, et al. (2017) Comprehensive attenuation of IL-25-induced airway hyperresponsiveness, inflammation and remodelling by the PI3K inhibitor LY294002. Respirology 22: 78–85. |
| [141] |
Welle S, Burgess K, Mehta S (2009) Stimulation of skeletal muscle myofibrillar protein synthesis, p70 S6 kinase phosphorylation, and ribosomal protein S6 phosphorylation by inhibition of myostatin in mature mice. Am J Physiol Endocrinol Metab 296: E567–572. doi: 10.1152/ajpendo.90862.2008
|
| [142] |
Jefferies HB, Fumagalli S, Dennis PB, et al. (1997) Rapamycin suppresses 5' TOP mRNA translation through inhibition of p70 s6k. EMBO J 16: 3693–3704. doi: 10.1093/emboj/16.12.3693
|
| [143] | Beretta L, Gingras AC, Svitkin YV, et al. (1996) Rapamycin blocks the phosphorylation of 4E-BP1 and inhibits cap-dependent initiation of translation. EMBO J 15: 658–664. |
| [144] |
Sneddon AA, Delday MI, Steven J, et al. (2001) Elevated IGF-II mRNA and phosphorylation of 4E-BP1 and p70S6k in muscle showing clenbuterol-induced anabolism. Am J Physiol Endocrinol Metab 281: E676–E682. doi: 10.1152/ajpendo.2001.281.4.E676
|
| [145] |
Bodine SC, Latres E, Baumhueter S, et al. (2001) Identification of ubiquitin ligases required for skeletal muscle atrophy. Science 294: 1704–1708. doi: 10.1126/science.1065874
|
| [146] |
Kline WO, Panaro FJ, Yang H, et al. (2007) Rapamycin inhibits the growth and muscle-sparing effects of clenbuterol. J Appl Physiol (1985) 102: 740–747. doi: 10.1152/japplphysiol.00873.2006
|
| [147] |
Okumura S, Fujita T, Cai W, et al. (2014) Epac1-dependent phospholamban phosphorylation mediates the cardiac response to stresses. J Clin Invest 124: 2785–2801. doi: 10.1172/JCI64784
|
| [148] |
Ohnuki Y, Umeki D, Mototani Y, et al. (2014) Role of cyclic AMP sensor Epac1 in masseter muscle hypertrophy and myosin heavy chain transition induced by beta2-adrenoceptor stimulation. J Physiol 592: 5461–5475. doi: 10.1113/jphysiol.2014.282996
|
| [149] | Ohnuki Y, Umeki D, Mototani Y, et al. (2016) Role of phosphodiesterase 4 expression in the Epac1 signaling-dependent skeletal muscle hypertrophic action of clenbuterol. Physiol Rep 4. |
| [150] |
Burnett DD, Paulk CB, Tokach MD, et al. (2016) Effects of Added Zinc on Skeletal Muscle Morphometrics and Gene Expression of Finishing Pigs Fed Ractopamine-HCL. Anim Biotechnol 27: 17–29. doi: 10.1080/10495398.2015.1069301
|
| [151] |
Py G, Ramonatxo C, Sirvent P, et al. (2015) Chronic clenbuterol treatment compromises force production without directly altering skeletal muscle contractile machinery. J Physiol 593: 2071–2084. doi: 10.1113/jphysiol.2014.287060
|
| [152] |
Rena G, Guo S, Cichy SC, et al. (1999) Phosphorylation of the transcription factor forkhead family member FKHR by protein kinase B. J Biol Chem 274: 17179–17183. doi: 10.1074/jbc.274.24.17179
|
| [153] |
Biggs WH, 3rd, Meisenhelder J, Hunter T, et al. (1999) Protein kinase B/Akt-mediated phosphorylation promotes nuclear exclusion of the winged helix transcription factor FKHR1. Proc Natl Acad Sci U S A 96: 7421–7426. doi: 10.1073/pnas.96.13.7421
|
| [154] |
Umeki D, Ohnuki Y, Mototani Y, et al. (2015) Protective Effects of Clenbuterol against Dexamethasone-Induced Masseter Muscle Atrophy and Myosin Heavy Chain Transition. PLoS One 10: e0128263. doi: 10.1371/journal.pone.0128263
|
| [155] |
de Palma L, Marinelli M, Pavan M, et al. (2008) Ubiquitin ligases MuRF1 and MAFbx in human skeletal muscle atrophy. Joint Bone Spine 75: 53–57. doi: 10.1016/j.jbspin.2007.04.019
|
| [156] |
Gomez-SanMiguel AB, Gomez-Moreira C, Nieto-Bona MP, et al. (2016) Formoterol decreases muscle wasting as well as inflammation in the rat model of rheumatoid arthritis. Am J Physiol Endocrinol Metab 310: E925–937. doi: 10.1152/ajpendo.00503.2015
|
| [157] |
Jesinkey SR, Korrapati MC, Rasbach KA, et al. (2014) Atomoxetine prevents dexamethasone-induced skeletal muscle atrophy in mice. J Pharmacol Exp Ther 351: 663–673. doi: 10.1124/jpet.114.217380
|
| [158] |
Yang YT, McElligott MA (1989) Multiple actions of beta-adrenergic agonists on skeletal muscle and adipose tissue. Biochem J 261: 1–10. doi: 10.1042/bj2610001
|
| [159] |
Peterla TA, Scanes CG (1990) Effect of beta-adrenergic agonists on lipolysis and lipogenesis by porcine adipose tissue in vitro. J Anim Sci 68: 1024–1029. doi: 10.2527/1990.6841024x
|
| [160] | Mayr B, Montminy M (2001) Transcriptional regulation by the phosphorylation-dependent factor CREB. Nat Rev Mol Cell Biol 2: 599–609. |
| [161] |
Pearen MA, Myers SA, Raichur S, et al. (2008) The orphan nuclear receptor, NOR-1, a target of beta-adrenergic signaling, regulates gene expression that controls oxidative metabolism in skeletal muscle. Endocrinology 149: 2853–2865. doi: 10.1210/en.2007-1202
|
| [162] |
Martinez JA, Portillo MP, Larralde J (1991) Anabolic actions of a mixed beta-adrenergic agonist on nitrogen retention and protein turnover. Horm Metab Res 23: 590–593. doi: 10.1055/s-2007-1003762
|
| [163] |
Watkins LE, Jones DJ, Mowrey DH, et al. (1990) The Effect of Various Levels of Ractopamine Hydrochloride on the Performance and Carcass Characteristics of Finishing Swine. J Anim Sci 68: 3588–3595. doi: 10.2527/1990.68113588x
|
| [164] |
Crome PK, McKeith FK, Carr TR, et al. (1996) Effect of ractopamine on growth performance, carcass composition, and cutting yields of pigs slaughtered at 107 and 125 kilograms. J Anim Sci 74: 709–716. doi: 10.2527/1996.744709x
|
| [165] |
Adams GR, Haddad F (1996) The relationships among IGF-1, DNA content, and protein accumulation during skeletal muscle hypertrophy. J Applied Physiol 81: 2509–2516. doi: 10.1152/jappl.1996.81.6.2509
|
| [166] |
Adams GR (2002) Invited Review: Autocrine/paracrine IGF-I and skeletal muscle adaptation. J Appl Physiol 93: 1159–1167. doi: 10.1152/japplphysiol.01264.2001
|
| [167] |
Grounds MD (2002) Reasons for the degeneration of ageing skeletal muscle: a central role for IGF-1 signalling. Biogerontology 3: 19–24. doi: 10.1023/A:1015234709314
|
| [168] |
Kicman AT, Miell JP, Teale JD, et al. (1997) Serum IGF-I and IGF binding proteins 2 and 3 as potential markers of doping with human GH. Clin Endocrinol 47: 43–50. doi: 10.1046/j.1365-2265.1997.2111036.x
|
| [169] |
Bahl N, Stone G, McLean M, et al. (2018) Decorin, a growth hormone-regulated protein in humans. Eur J Endocrinol 178: 147–154. doi: 10.1530/EJE-17-0844
|
| [170] |
Kanzleiter T, Rath M, Gorgens SW, et al. (2014) The myokine decorin is regulated by contraction and involved in muscle hypertrophy. Biochem Biophys Res Commun 450: 1089–1094. doi: 10.1016/j.bbrc.2014.06.123
|
| [171] |
Musaro A, McCullagh K, Paul A, et al. (2001) Localized Igf-1 transgene expression sustains hypertrophy and regeneration in senescent skeletal muscle. Nat Genet 27: 195–200. doi: 10.1038/84839
|
| [172] |
Adams GR, McCue SA (1998) Localized infusion of IGF-I results in skeletal muscle hypertrophy in rats. J Appl Physiol 84: 1716–1722. doi: 10.1152/jappl.1998.84.5.1716
|
| [173] |
Yin HN, Chai JK, Yu YM, et al. (2009) Regulation of signaling pathways downstream of IGF-I/insulin by androgen in skeletal muscle of glucocorticoid-treated rats. J Trauma 66: 1083–1090. doi: 10.1097/TA.0b013e31817e7420
|
| [174] | Jang YJ, Son HJ, Choi YM, et al. (2017) Apigenin enhances skeletal muscle hypertrophy and myoblast differentiation by regulating Prmt7. Oncotarget 8: 78300–78311. |
| [175] |
Song YH, Li Y, Du J, et al. (2005) Muscle-specific expression of IGF-1 blocks angiotensin II-induced skeletal muscle wasting. J Clin Invest 115: 451–458. doi: 10.1172/JCI22324
|
| [176] |
LaVigne EK, Jones AK, Londono AS, et al. (2015) Muscle growth in young horses: Effects of age, cytokines, and growth factors. J Anim Sci 93: 5672–5680. doi: 10.2527/jas.2015-9634
|
| [177] |
Chin ER, Olson EN, Richardson JA, et al. (1998) A calcineurin-dependent transcriptional pathway controls skeletal muscle fiber type. Genes Dev 12: 2499–2509. doi: 10.1101/gad.12.16.2499
|
| [178] |
Delling U, Tureckova J, Lim HW, et al. (2000) A calcineurin-NFATc3-dependent pathway regulates skeletal muscle differentiation and slow myosin heavy-chain expression. Mol Cell Biol 20: 6600–6611. doi: 10.1128/MCB.20.17.6600-6611.2000
|
| [179] |
Semsarian C, Wu MJ, Ju YK, et al. (1999) Skeletal muscle hypertrophy is mediated by a Ca2+-dependent calcineurin signalling pathway. Nature 400: 576–581. doi: 10.1038/23054
|
| [180] |
Miyashita T, Takeishi Y, Takahashi H, et al. (2001) Role of calcineurin in insulin-like growth factor-1-induced hypertrophy of cultured adult rat ventricular myocytes. Jpn Circ J 65: 815–819. doi: 10.1253/jcj.65.815
|
| [181] |
Musaro A, McCullagh KJA, Naya FJ, et al. (1999) IGF-1 induces skeletal myocyte hypertrophy through calcineurin in association with GATA-2 and NF-ATc1. Nature 400: 581–585. doi: 10.1038/23060
|
| [182] |
Swoap SJ, Hunter RB, Stevenson EJ, et al. (2000) The calcineurin-NFAT pathway and muscle fiber-type gene expression. Am J Physiol Cell Physiol 279: C915–C924. doi: 10.1152/ajpcell.2000.279.4.C915
|
| [183] |
White TA, LeBrasseur NK (2014) Myostatin and sarcopenia: opportunities and challenges - a mini-review. Gerontology 60: 289–293. doi: 10.1159/000356740
|
| [184] |
Patel K, Amthor H (2005) The function of Myostatin and strategies of Myostatin bloackde - new hope for therapies aimed at promoting growth of skeletal muscle. Neuromuscular Disord 15: 117–126. doi: 10.1016/j.nmd.2004.10.018
|
| [185] |
Sartori R, Milan G, Patron M, et al. (2009) Smad2 and 3 transcription factors control muscle mass in adulthood. Am J Physiol Cell Physiol 296: C1248–1257. doi: 10.1152/ajpcell.00104.2009
|
| [186] |
Smith RC, Lin BK (2013) Moystatin inhibitors as therapies for muscle wasting associated with cancer and other disorders. Curr Opin Support Palliat Care 7: 352–360. doi: 10.1097/SPC.0000000000000013
|
| [187] |
Winbanks CE, Weeks KL, Thomson RE, et al. (2012) Follistatin-mediated skeletal muscle hypertrophy is regulated by Smad3 and mTOR independently of myostatin. J Cell Biol 197: 997–1008. doi: 10.1083/jcb.201109091
|
| [188] |
Winbanks CE, Chen JL, Qian H, et al. (2013) The bone morphogenetic protein axis is a positive regulator of skeletal muscle mass. J Cell Biol 203: 345–357. doi: 10.1083/jcb.201211134
|
| [189] |
Barbe C, Kalista S, Loumaye A, et al. (2015) Role of IGF-I in follistatin-induced skeletal muscle hypertrophy. Am J Physiol Endocrinol Metab 309: E557–567. doi: 10.1152/ajpendo.00098.2015
|
| [190] |
Wang Q, Guo T, Portas J, et al. (2015) A soluble activin receptor type IIB does not improve blood glucose in streptozotocin-treated mice. Int J Biol Sci 11: 199–208. doi: 10.7150/ijbs.10430
|
| [191] |
Barreiro E, Tajbakhsh S (2017) Epigenetic regulation of muscle development. J Muscle Res Cell Motil 38: 31–35. doi: 10.1007/s10974-017-9469-5
|
| [192] |
Grazioli E, Dimauro I, Mercatelli N, et al. (2017) Physical activity in the prevention of human diseases: role of epigenetic modifications. BMC Genomics 18: 802. doi: 10.1186/s12864-017-4193-5
|
| [193] |
Saccone V, Puri PL (2010) Epigenetic regulation of skeletal myogenesis. Organogenesis 6: 48–53. doi: 10.4161/org.6.1.11293
|
| [194] |
Sincennes MC, Brun CE, Rudnicki MA (2016) Concise Review: Epigenetic Regulation of Myogenesis in Health and Disease. Stem Cells Transl Med 5: 282–290. doi: 10.5966/sctm.2015-0266
|
| [195] |
Townley-Tilson WH, Callis TE, Wang D (2010) MicroRNAs 1, 133, and 206: critical factors of skeletal and cardiac muscle development, function, and disease. Int J Biochem Cell Biol 42: 1252–1255. doi: 10.1016/j.biocel.2009.03.002
|
| [196] |
Proctor CJ, Goljanek-Whysall K (2017) Using computer simulation models to investigate the most promising microRNAs to improve muscle regeneration during ageing. Sci Rep 7: 12314. doi: 10.1038/s41598-017-12538-6
|
| [197] |
Yu H, Lu Y, Li Z, et al. (2014) microRNA-133: expression, function and therapeutic potential in muscle diseases and cancer. Curr Drug Targets 15: 817–828. doi: 10.2174/1389450115666140627104151
|
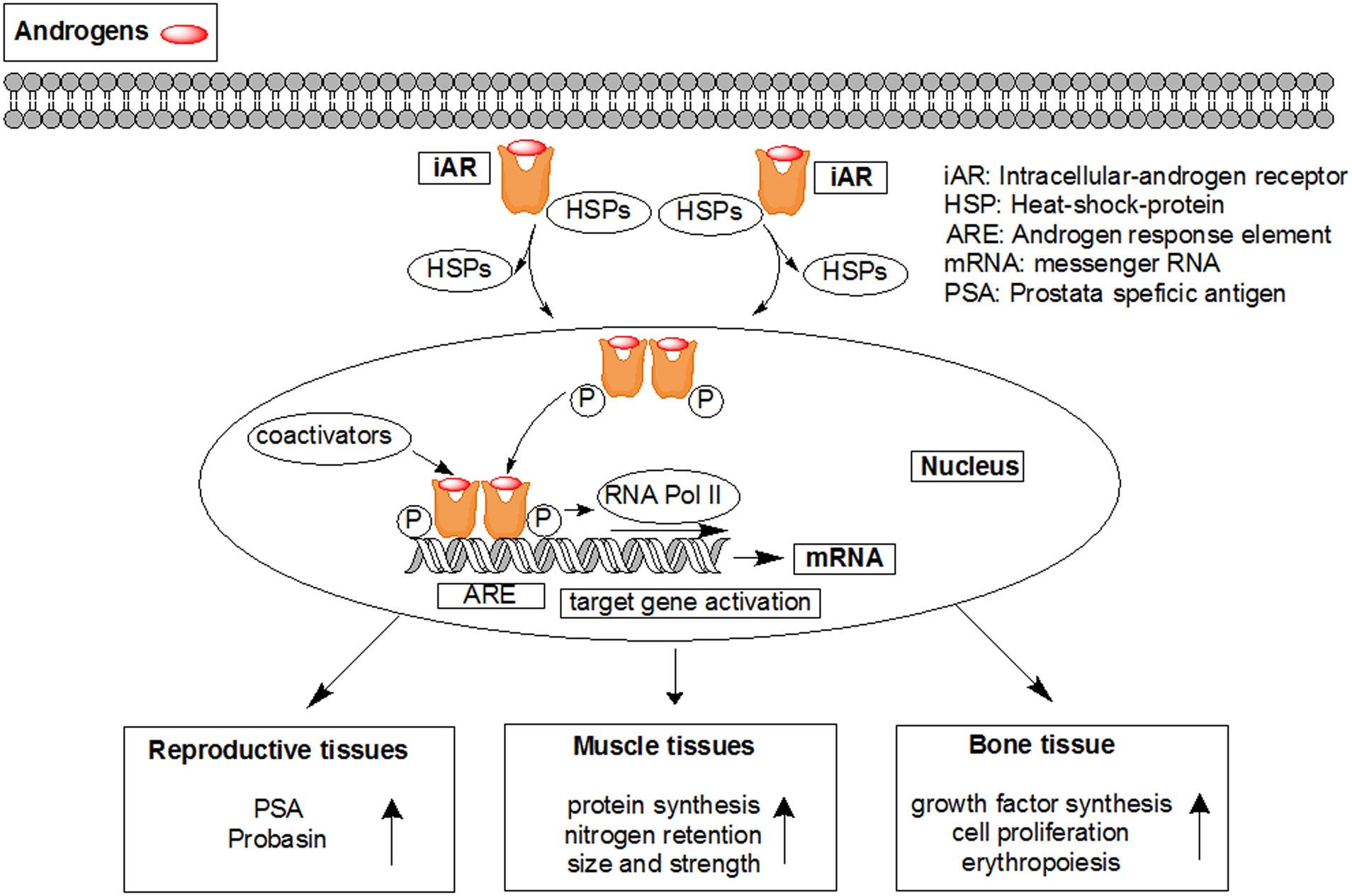
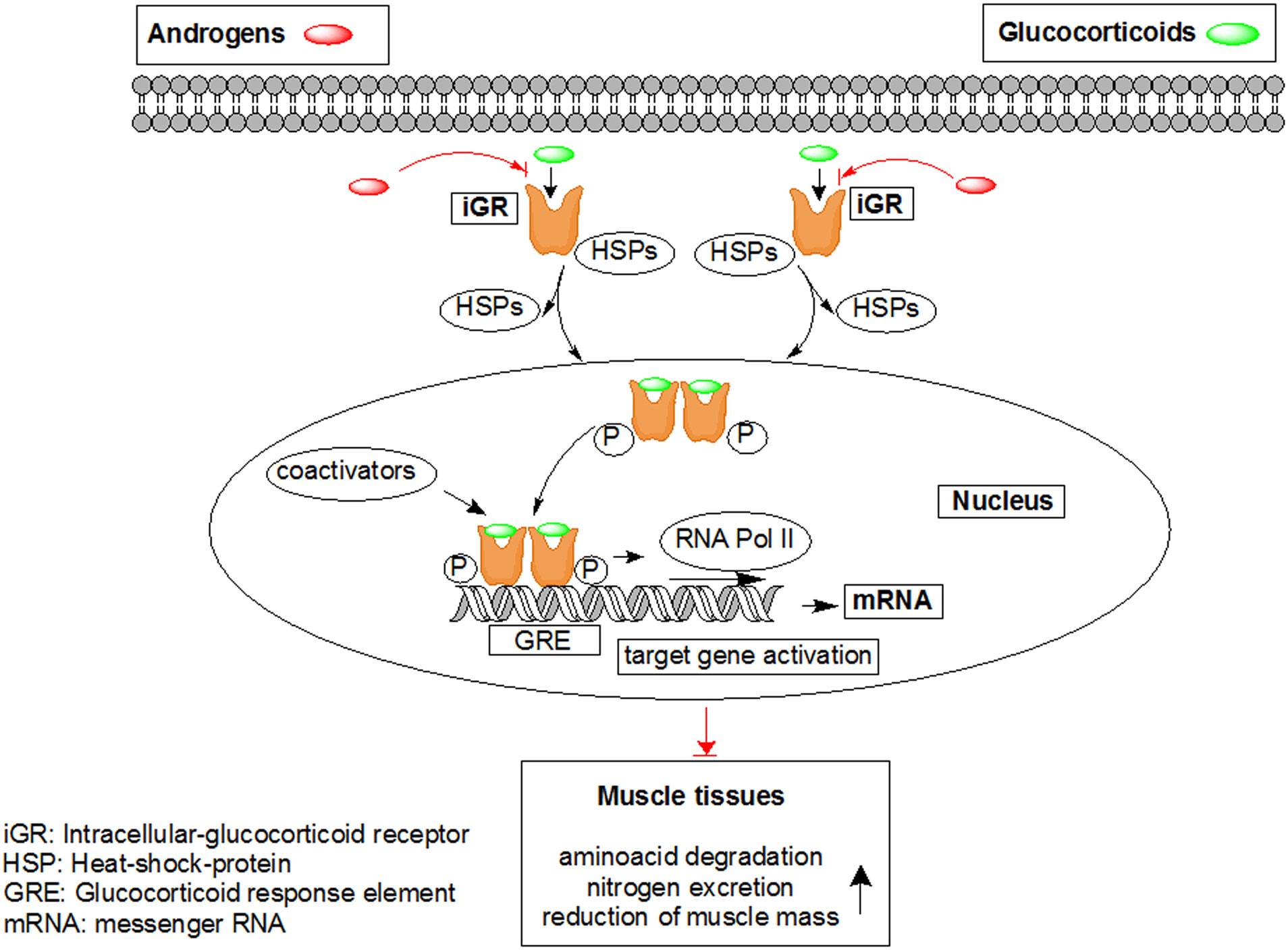
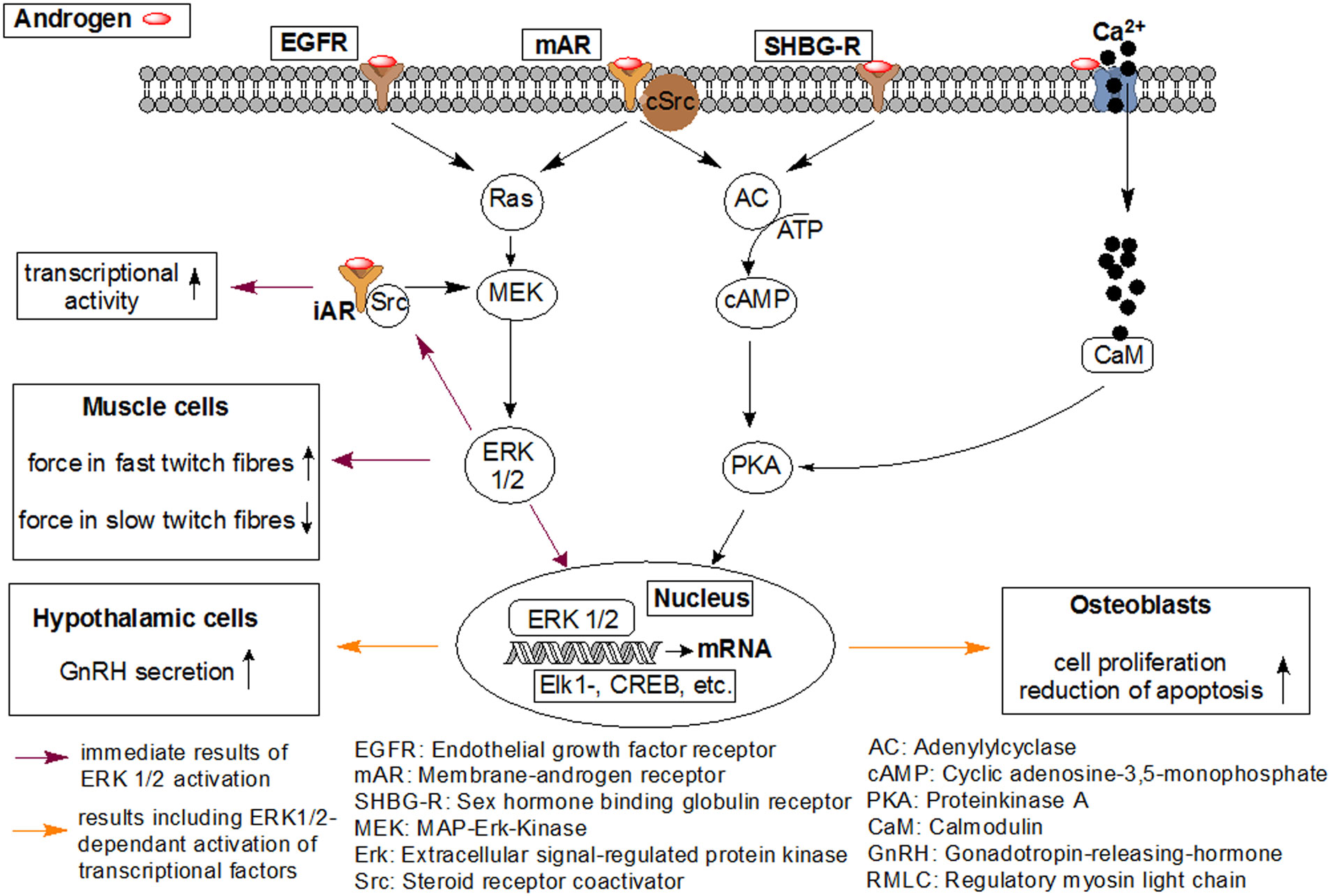
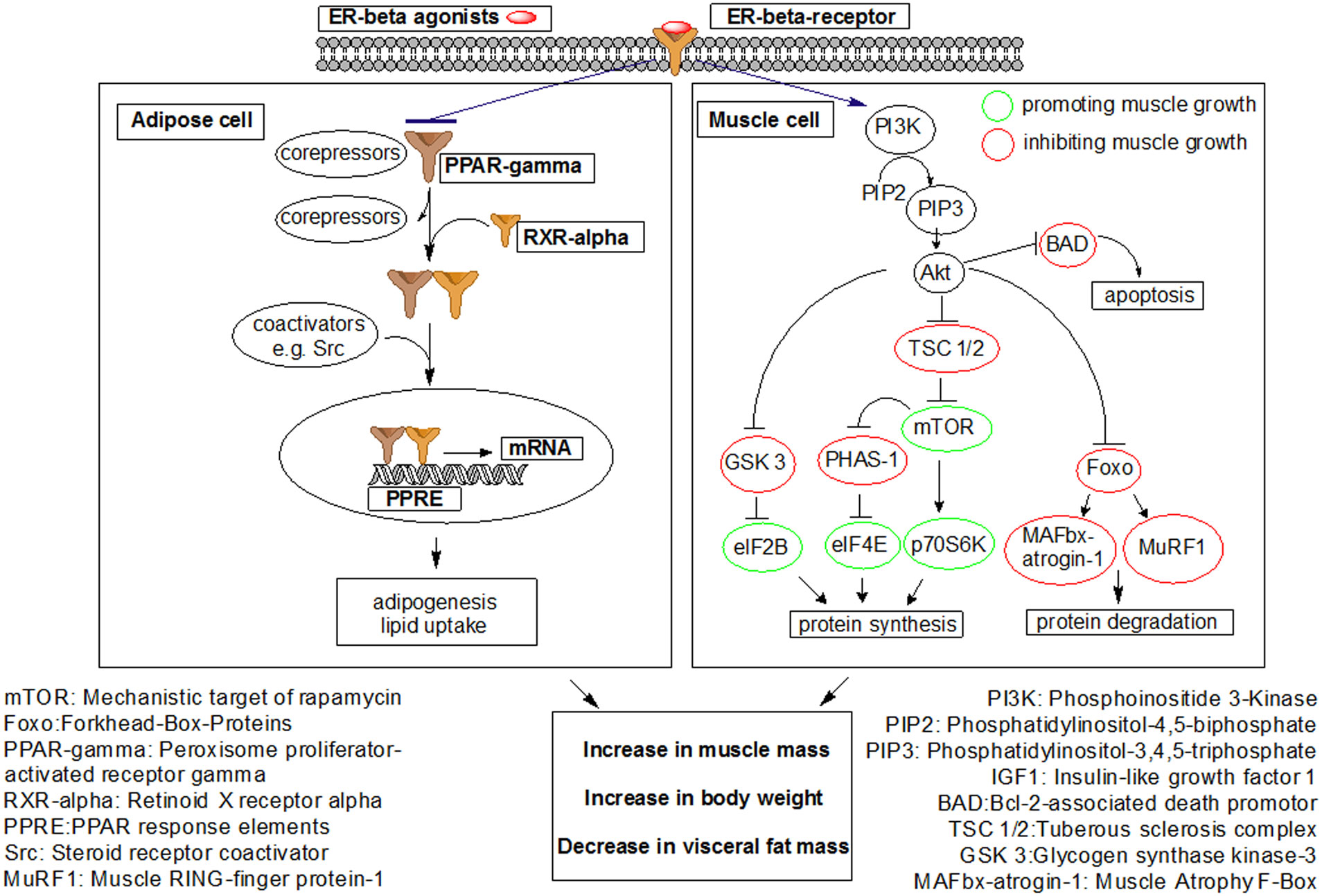
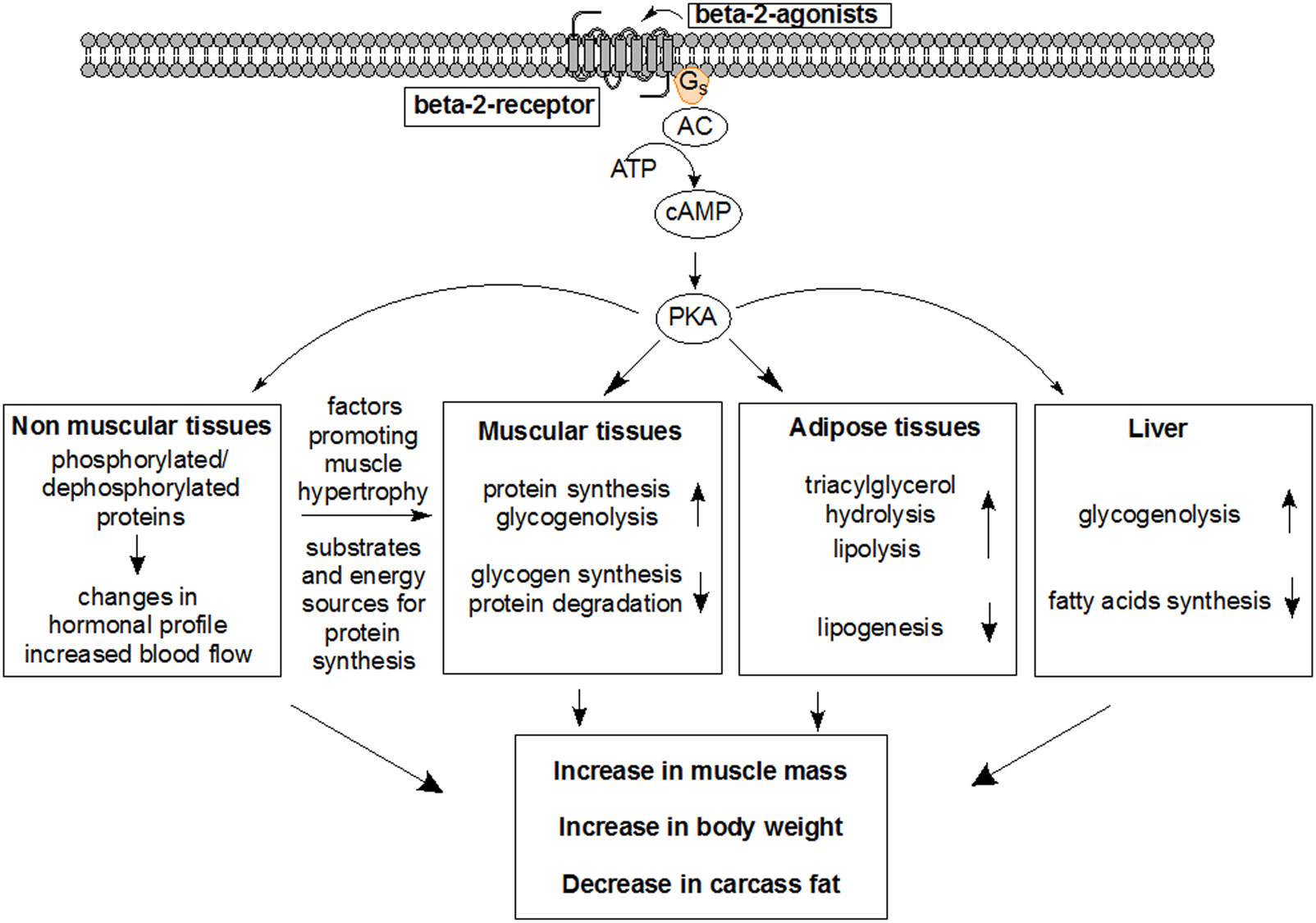
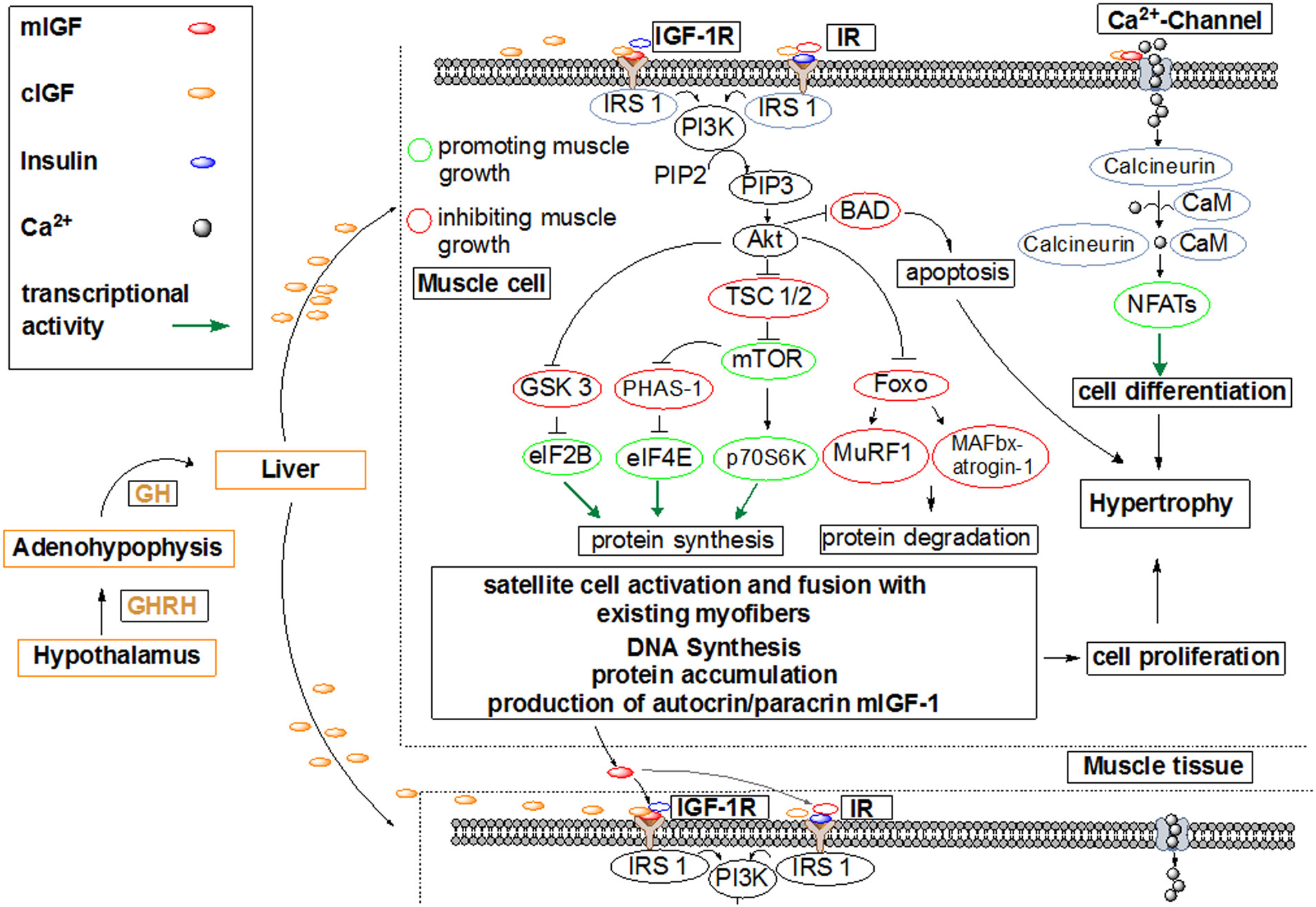
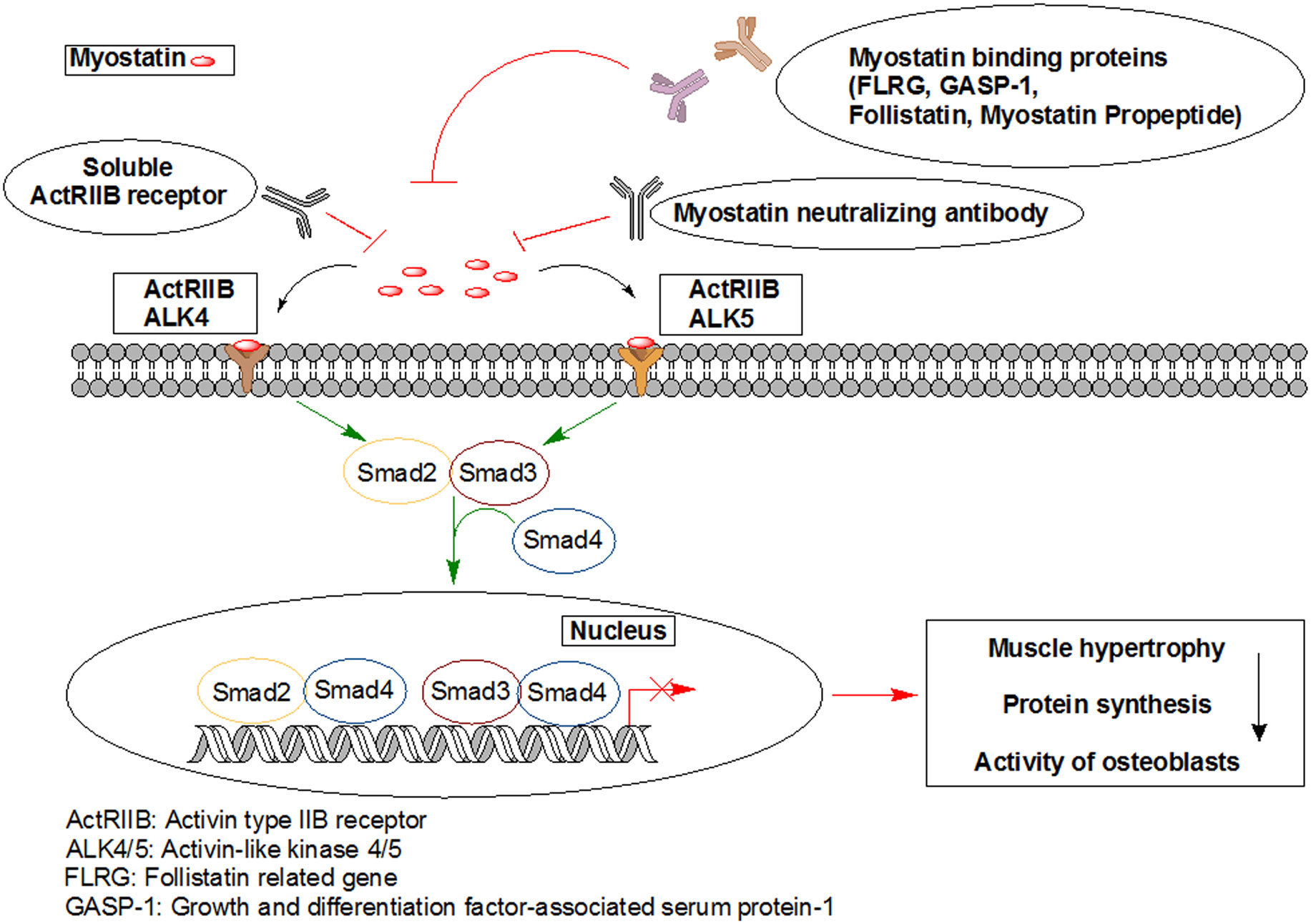
Figures(7)

Maria Kristina Parr, Anna Müller-Schöll. Pharmacology of doping agents—mechanisms promoting muscle hypertrophy[J]. AIMS Molecular Science, 2018, 5(2): 131-159. doi: 10.3934/molsci.2018.2.131







 DownLoad:
DownLoad: