Citation: Mohammad Shamim Ahasan, Thomas B. Waltzek, Leigh Owens, Ellen Ariel. Characterisation and comparison of the mucosa-associated bacterial communities across the gastrointestinal tract of stranded green turtles, Chelonia mydas[J]. AIMS Microbiology, 2020, 6(4): 361-378. doi: 10.3934/microbiol.2020022

| [1] |
Chung H, Kasper DL (2010) Microbiota-stimulated immune mechanisms to maintain gut homeostasis. Curr Opin Immunol 22: 455-460. doi: 10.1016/j.coi.2010.06.008

|
| [2] |
Dethlefsen L, McFall-Ngai M, Relman DA (2007) An ecological and evolutionary perspective on human-microbe mutualism and disease. Nature 449. doi: 10.1038/nature06245
|
| [3] |
Gill SR, Pop M, DeBoy RT, et al. (2006) Metagenomic analysis of the human distal gut microbiome. Science 312: 1355-1359. doi: 10.1126/science.1124234
|
| [4] |
Sartor RB, Mazmanian SK (2012) Intestinal microbes in inflammatory bowel diseases. Am J Gastroenterol Suppl 1: 15-21. doi: 10.1038/ajgsup.2012.4
|
| [5] |
Sobhani I, Tap J, Roudot-Thoraval F, et al. (2011) Microbial dysbiosis in colorectal cancer (CRC) patients. PloS one 6: e16393. doi: 10.1371/journal.pone.0016393
|
| [6] |
MacFarlane GT, Macfarlane L (2009) Acquisition, evolution and maintenance of the normal gut microbiota. Dig Dis 27: 90-98. doi: 10.1159/000268127
|
| [7] |
Vaarala O (2011) The gut as a regulator of early inflammation in type 1 diabetes. Curr Opin Endocrinol Diabetes Obes 18: 241-247. doi: 10.1097/MED.0b013e3283488218
|
| [8] |
Power SE, O'Toole PW, Stanton C, et al. (2014) Intestinal microbiota, diet and health. Br J Nutr 111: 387-402. doi: 10.1017/S0007114513002560
|
| [9] |
MM OD, Harris HM, Jeffery IB, et al. (2013) The core faecal bacterial microbiome of Irish Thoroughbred racehorses. Lett Appl Microbiol 57: 492-501. doi: 10.1111/lam.12137
|
| [10] |
Benson AK, Kelly SA, Legge R, et al. (2010) Individuality in gut microbiota composition is a complex polygenic trait shaped by multiple environmental and host genetic factors. Proc Natl Acad Sci 107: 18933-18938. doi: 10.1073/pnas.1007028107
|
| [11] |
Sommer F, Backhed F (2013) The gut microbiota--masters of host development and physiology. Nat Rev Microbiol 11: 227-238. doi: 10.1038/nrmicro2974
|
| [12] |
Carey HV, Walters WA, Knight R (2013) Seasonal restructuring of the ground squirrel gut microbiota over the annual hibernation cycle. Am J Physiol Regul Integr Comp Physiol 304: R33-R42. doi: 10.1152/ajpregu.00387.2012
|
| [13] |
MacFariane RD, McLaughlin JJ, Bullock G (1986) Quantitative and qualitative studies of gut flora in striped bass from estuarine and coastal marine environments. J wildl dis 22: 344-348. doi: 10.7589/0090-3558-22.3.344
|
| [14] |
Sullam KE, Essinger SD, Lozupone CA, et al. (2012) Environmental and ecological factors that shape the gut bacterial communities of fish: a meta-analysis. Mol Ecol 21: 3363-3378. doi: 10.1111/j.1365-294X.2012.05552.x
|
| [15] |
Mao S, Zhang M, Liu J, et al. (2015) Characterising the bacterial microbiota across the gastrointestinal tracts of dairy cattle: membership and potential function. Sci Rep 5: 16116. doi: 10.1038/srep16116
|
| [16] |
Nelson TM, Rogers TL, Brown MV (2013) The gut bacterial community of mammals from marine and terrestrial habitats. PLoS One 8: e83655. doi: 10.1371/journal.pone.0083655
|
| [17] |
Li T, Long M, Gatesoupe F-J, et al. (2015) Comparative Analysis of the Intestinal Bacterial Communities in Different Species of Carp by Pyrosequencing. Microb Ecol 69: 25-36. doi: 10.1007/s00248-014-0480-8
|
| [18] |
Wang W, Cao J, Li J-R, et al. (2016) Comparative analysis of the gastrointestinal microbial communities of bar-headed goose (Anser indicus) in different breeding patterns by high-throughput sequencing. Microbiol Res 182: 59-67. doi: 10.1016/j.micres.2015.10.003
|
| [19] |
Wilkinson N, Hughes RJ, Aspden WJ, et al. (2016) The gastrointestinal tract microbiota of the Japanese quail, Coturnix japonica. Appl Microbiol Biotechnol 100: 4201-4209. doi: 10.1007/s00253-015-7280-z
|
| [20] |
Yang Y, Deng Y, Cao L (2016) Characterising the interspecific variations and convergence of gut microbiota in Anseriformes herbivores at wintering areas. Sci Rep 6: 32655. doi: 10.1038/srep32655
|
| [21] |
Li K, Guan W, Wei G, et al. (2007) Phylogenetic analysis of intestinal bacteria in the Chinese mitten crab (Eriocheir sinensis). J Appl Microbiol 103: 675-682. doi: 10.1111/j.1365-2672.2007.03295.x
|
| [22] |
Li L, Yan B, Li S, et al. (2016) A comparison of bacterial community structure in seawater pond with shrimp, crab, and shellfish cultures and in non-cultured pond in Ganyu, Eastern China. Ann Microbiol 66: 317-328. doi: 10.1007/s13213-015-1111-4
|
| [23] |
Souza DT, Genuário DB, Silva FSP, et al. (2016) Analysis of bacterial composition in marine sponges reveals the influence of host phylogeny and environment. FEMS Microbiol Ecol 93: fiw204-fiw204. doi: 10.1093/femsec/fiw204
|
| [24] |
Price JT, Paladino FV, Lamont MM, et al. (2017) Characterization of the juvenile green turtle (Chelonia mydas) microbiome throughout an ontogenetic shift from pelagic to neritic habitats. PloS one 12: e0177642. doi: 10.1371/journal.pone.0177642
|
| [25] |
Ahasan MS, Waltzek TB, Huerlimann R, et al. (2017) Fecal bacterial communities of wild-captured and stranded green turtles (Chelonia mydas) on the Great Barrier Reef. FEMS Microbiol Ecol 93: fix139. doi: 10.1093/femsec/fix139
|
| [26] |
McDermid KJ, Kittle III RP, Veillet A, et al. (2020) Identification of Gastrointestinal Microbiota in Hawaiian Green Turtles (Chelonia mydas). Evol Bioinf Online 16: 1176934320914603. doi: 10.1177/1176934320914603
|
| [27] | Bjorndal KA, Lutz P, Musick J (1997) Foraging ecology and nutrition of sea turtles. Biol sea turtles 1: 199-231. |
| [28] |
Bjorndal LA (1979) Cellulose digestion and volatile fatty-acid production in the green turtle Chelonia mydas. Comp Biochem Physiol 63: 127-134. doi: 10.1016/0300-9629(79)90638-8
|
| [29] | Bjorndal KA, Suganuma H, Bolten AB (1991) Digestive fermentation in green turtles, Chelonia mydas, feeding on algae. B Mar Sci 48: 66-71. |
| [30] |
Costa MC, Arroyo LG, Allen-Vercoe E, et al. (2012) Comparison of the fecal microbiota of healthy horses and horses with colitis by high throughput sequencing of the V3-V5 region of the 16S rRNA gene. PLoS One 7: e41484. doi: 10.1371/journal.pone.0041484
|
| [31] |
Guarner F, Malagelada JR (2003) Gut flora in health and disease. Lancet 361: 512-519. doi: 10.1016/S0140-6736(03)12489-0
|
| [32] |
Ahasan MS, Waltzek TB, Huerlimann R, et al. (2018) Comparative analysis of gut bacterial communities of green turtles (Chelonia mydas) pre-hospitalization and post-rehabilitation by high-throughput sequencing of bacterial 16S rRNA gene. Microbiol Res 207: 91-99. doi: 10.1016/j.micres.2017.11.010
|
| [33] |
Bloodgood JC, Hernandez SM, Isaiah A, et al. (2020) The effect of diet on the gastrointestinal microbiome of juvenile rehabilitating green turtles (Chelonia mydas). PLOS One 15: e0227060. doi: 10.1371/journal.pone.0227060
|
| [34] |
Morais RA, Dos Santos RG, Longo GO, et al. (2014) Direct evidence for gradual ontogenetic dietary shift in the green turtle, Chelonia mydas. Chelonian Conserv Biol 13: 260-266. doi: 10.2744/CCB-1058.1
|
| [35] | McNally KL (2020) Characterization of Microbial Communities Across Disease States and Environmental Conditions in Kemp's Ridley (Lepidochelys Kempii) and Green Sea Turtles (Chelonia Mydas). Graduate Doctoral Dissertations 551. |
| [36] | Li D, Chen H, Mao B, et al. (2017) Microbial biogeography and core microbiota of the Rat digestive tract. Sci Rep 8. |
| [37] |
Neumann LM, Dehority BA (2008) An investigation of the relationship between fecal and rumen bacterial concentrations in sheep. Zoo Biol 27: 100-108. doi: 10.1002/zoo.20166
|
| [38] |
Eckburg P, Bik E, Bernstein C, et al. (2005) Diversity of the human intestinal microbial flora. Science 308: 1635-1638. doi: 10.1126/science.1110591
|
| [39] |
Kelly J, Daly K, Moran AW, et al. (2016) Composition and diversity of mucosa–associated microbiota along the entire length of the pig gastrointestinal tract; dietary influences. Environ Microbiol 19: 1425-1438. doi: 10.1111/1462-2920.13619
|
| [40] |
Daly K, Stewart CS, Flint HJ, et al. (2001) Bacterial diversity within the equine large intestine as revealed by molecular analysis of cloned 16S rRNA genes. FEMS Microbiol Ecol 38: 141-151. doi: 10.1111/j.1574-6941.2001.tb00892.x
|
| [41] | (2013) DEHPDepartment of Environment and Heritage Protection. Monitoring and Sampling Manual 2009, Version 2, July 2013, format edits ed. . Australia: Department of Environment and Heritage Protection, QLD. |
| [42] |
Caporaso JG, Kuczynski J, Stombaugh J, et al. (2010) QIIME allows analysis of high-throughput community sequencing data. Nat Methods 7: 335-336. doi: 10.1038/nmeth.f.303
|
| [43] |
Masella AP, Bartram AK, Truszkowski JM, et al. (2012) PANDAseq: paired-end assembler for illumina sequences. BMC Bioinformatics 13: 31. doi: 10.1186/1471-2105-13-31
|
| [44] |
Edgar RC, Haas BJ, Clemente JC, et al. (2011) UCHIME improves sensitivity and speed of chimera detection. Bioinformatics 27: 2194-2200. doi: 10.1093/bioinformatics/btr381
|
| [45] |
McMurdie PJ, Holmes S (2013) phyloseq: An R package for reproducible interactive analysis and graphics of microbiome census data. PLOS One 8: e61217. doi: 10.1371/journal.pone.0061217
|
| [46] |
Lozupone C, Knight R (2005) UniFrac: a new phylogenetic method for comparing microbial communities. Appl Environ Microbiol 8228-8235. doi: 10.1128/AEM.71.12.8228-8235.2005
|
| [47] | Zakrzewski M, Proietti C, Ellis JJ, et al. (2017) Calypso: a user-friendly web-server for mining and visualizing microbiome–environment interactions. Bioinformatics 33: 782-783. |
| [48] |
Bray JR, Curtis JT (1957) An ordination of the upland forest communities of Southern Wisconsin. Ecol Monogr 27: 325-349. doi: 10.2307/1942268
|
| [49] | Benjamini Y, Hochberg Y (1995) Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Stat Soc Series B 289-300. |
| [50] | Bjorndal KA (1995) Biology and conservation of sea turtles Washington: Smithsonian Institution Press. |
| [51] |
Lutz PL, Musick JA, Wyneken J (2002) The biology of sea turtles CRC press. doi: 10.1201/9781420040807
|
| [52] |
Pedrós-Alió C (2006) Marine microbial diversity: can it be determined? Trends Microbiol 14: 257-263. doi: 10.1016/j.tim.2006.04.007
|
| [53] |
Zinger L, Amaral-Zettler LA, Fuhrman JA, et al. (2011) Global patterns of bacterial beta-diversity in seafloor and seawater ecosystems. PLoS One 6: e24570. doi: 10.1371/journal.pone.0024570
|
| [54] |
Gu S, Chen D, Zhang JN, et al. (2013) Bacterial community mapping of the mouse gastrointestinal tract. PloS One 8: e74957. doi: 10.1371/journal.pone.0074957
|
| [55] |
Flint HJ, Scott KP, Louis P, et al. (2012) The role of the gut microbiota in nutrition and health. Nat Rev Gastroenterol Hepatol 9: 577-589. doi: 10.1038/nrgastro.2012.156
|
| [56] |
Byndloss MX, Olsan EE, Rivera-Chávez F, et al. (2017) Microbiota-activated PPAR-γ signaling inhibits dysbiotic Enterobacteriaceae expansion. Science 357: 570-575. doi: 10.1126/science.aam9949
|
| [57] |
Campos P, Guivernau M, Prenafeta-Boldú FX, et al. (2018) Fast acquisition of a polysaccharide fermenting gut microbiome by juvenile green turtles Chelonia mydas after settlement in coastal habitats. Microbiome 6: 69. doi: 10.1186/s40168-018-0454-z
|
| [58] |
Turnbaugh PJ, Bäckhed F, Fulton L, et al. (2008) Diet-induced obesity is linked to marked but reversible alterations in the mouse distal gut microbiome. Cell Host Microbe 3: 213-223. doi: 10.1016/j.chom.2008.02.015
|
| [59] |
Hong PY, Wheeler E, Cann IK, et al. (2011) Phylogenetic analysis of the fecal microbial community in herbivorous land and marine iguanas of the Galapagos Islands using 16S rRNA-based pyrosequencing. ISME J 5: 1461-1470. doi: 10.1038/ismej.2011.33
|
| [60] |
Arthur KE, Boyle MC, Limpus CJ (2008) Ontogenetic changes in diet and habitat use in green sea turtle (Chelonia mydas) life history. Marine Ecology Progress Series 362: 303-311. doi: 10.3354/meps07440
|
| [61] |
Kim M, Morrison M, Yu Z (2011) Status of the phylogenetic diversity census of ruminal microbiomes. FEMS Microbiol Ecol 76: 49-63. doi: 10.1111/j.1574-6941.2010.01029.x
|
| [62] |
Kim M, Kim J, Kuehn L, et al. (2014) Investigation of bacterial diversity in the feces of cattle fed different diets. J Anim Sci 92: 683-694. doi: 10.2527/jas.2013-6841
|
| [63] |
Mao S, Zhang R, Wang D, et al. (2012) The diversity of the fecal bacterial community and its relationship with the concentration of volatile fatty acids in the feces during subacute rumen acidosis in dairy cows. BMC Vet Res 8: 237. doi: 10.1186/1746-6148-8-237
|
| [64] |
Kashyap PC, Marcobal A, Ursell LK, et al. (2013) Complex interactions among diet, gastrointestinal transit, and gut microbiota in humanized mice. Gastroenterology 144: 967-977. doi: 10.1053/j.gastro.2013.01.047
|
| [65] |
Walker AW, Duncan SH, William Leitch EC, et al. (2005) pH and peptide supply can radically alter bacterial populations and short-chain fatty acid ratios within microbial communities from the human colon. Appl Environ Microbiol 71: 3692-3700. doi: 10.1128/AEM.71.7.3692-3700.2005
|
| [66] |
Bartram AK, Lynch MDJ, Stearns JC, et al. (2011) Generation of multimillion-sequence 16S rRNA gene libraries from complex microbial communities by assembling paired-end Illumina reads. Appl Environ Microbiol 77: 3846-3852. doi: 10.1128/AEM.02772-10
|
| [67] |
Savage DC (1977) Microbial ecology of the gastrointestinal tract. Annu Rev Microbiol 31: 107-133. doi: 10.1146/annurev.mi.31.100177.000543
|
| [68] | Bergey DH, Garrity GM (2004) Bergey's manual of systematic bacteriology: the proteobacteria Springer. |
| [69] |
Chow WL, Lee YK (2006) Mucosal interactions and gastrointestinal microbiota. Gastrointestinal Microbiology New York, NY: Taylor and Francis, 123-136. doi: 10.3109/9781420014952-7
|
| [70] | Wilson M (2005) Microbial inhabitants of humans: their ecology and role in health and disease Cambridge University Press. |
| [71] |
Amaro C, Biosca EG (1996) Vibrio vulnificus biotype 2, pathogenic for eels, is also an opportunistic pathogen for humans. Appl Environ Microbiol 62: 1454-1457. doi: 10.1128/AEM.62.4.1454-1457.1996
|
| [72] | Brenner DJ, Farmer J (1984) Enterobacteriaceae. Bergey's Manual of Systematics of Archaea and Bacteria . |
| [73] | Dworkin M, Falkow S, Rosenberg E, et al. (2006) Proteobacteria: Gamma Subclass. The Prokaryotes Springer Science & Business Media. |
| [74] |
Schaal K, Schofield G, Pulverer G (1980) Taxonomy and clinical significance of Actinomycetaceae and Propionibacteriaceae. Infection 8: S122-S130. doi: 10.1007/BF01639868
|
| [75] |
Stackebrandt E, Cummins CS, Johnson JL (2006) Family propionibacteriaceae: the genus Propionibacterium. The prokaryotes Springer, 400-418. doi: 10.1007/0-387-30743-5_19
|
| [76] |
Wang K, Lu W, Tu Q, et al. (2016) Preliminary analysis of salivary microbiome and their potential roles in oral lichen planus. Sci Rep 6: 22943. doi: 10.1038/srep22943
|
| [77] |
Flint HJ, Scott KP, Duncan SH, et al. (2012) Microbial degradation of complex carbohydrates in the gut. Gut Microbes 3: 289-306. doi: 10.4161/gmic.19897
|
| [78] |
Martens EC, Lowe EC, Chiang H, et al. (2011) Recognition and degradation of plant cell wall polysaccharides by two human gut symbionts. PLoS Biol 9: e1001221. doi: 10.1371/journal.pbio.1001221
|
| [79] |
Salyers A, Vercellotti J, West S, et al. (1977) Fermentation of mucin and plant polysaccharides by strains of Bacteroides from the human colon. Appl Environ Microbiol 33: 319-322. doi: 10.1128/AEM.33.2.319-322.1977
|
 microbiol-06-04-022-s001.pdf microbiol-06-04-022-s001.pdf |
 |
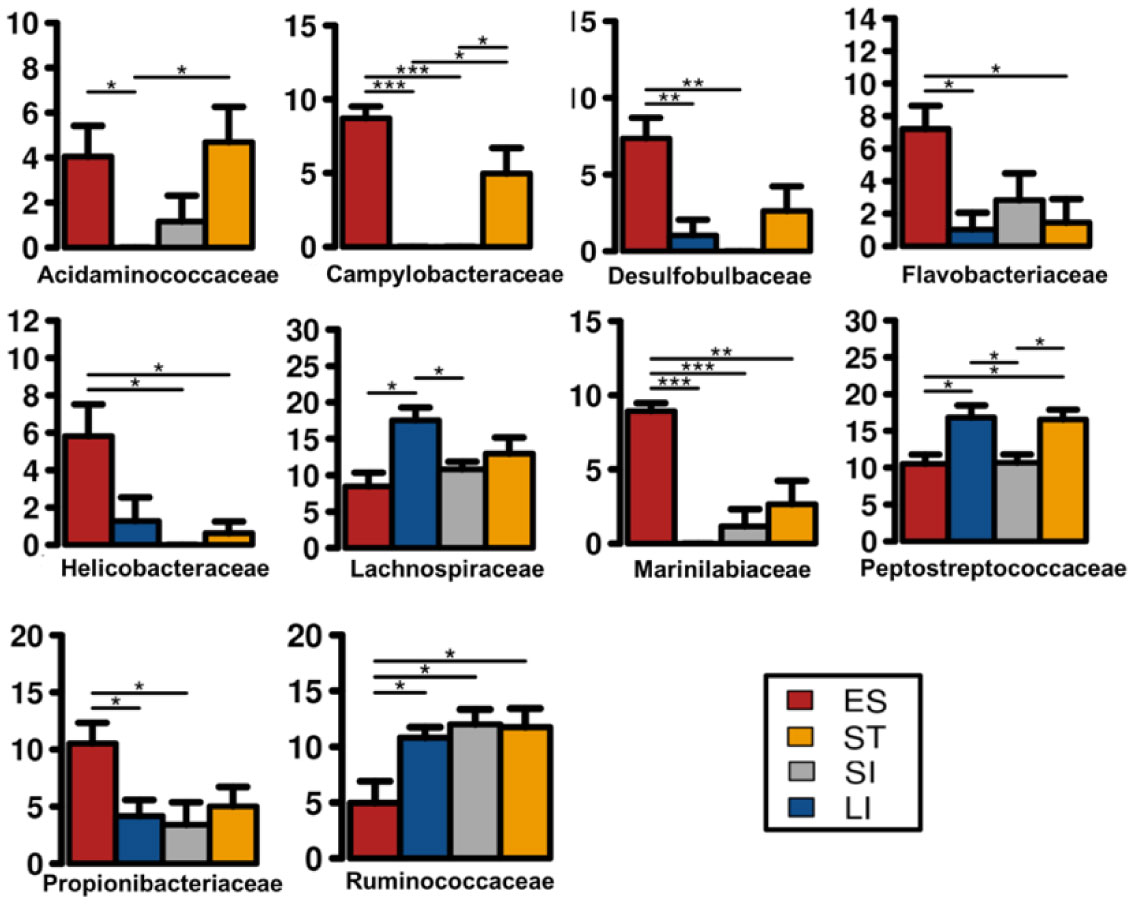
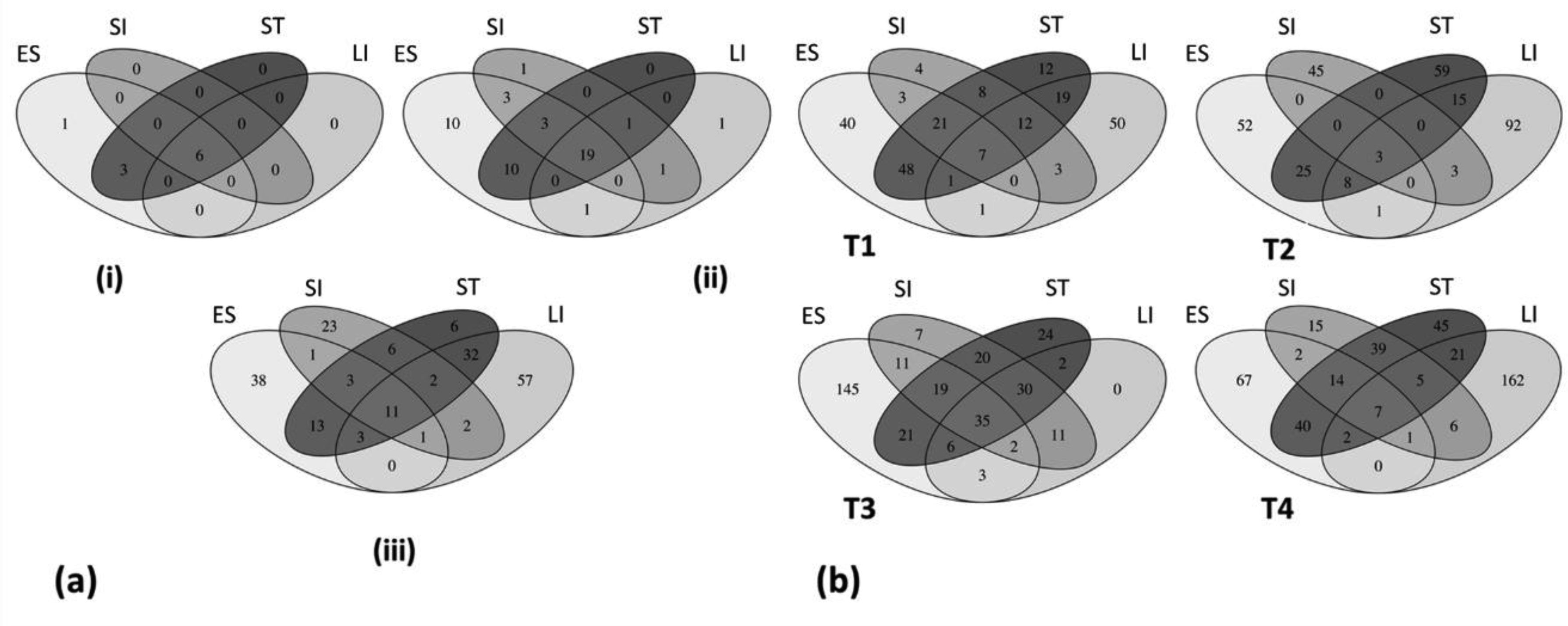
Figures(6) / Tables(1)

Mohammad Shamim Ahasan, Thomas B. Waltzek, Leigh Owens, Ellen Ariel. Characterisation and comparison of the mucosa-associated bacterial communities across the gastrointestinal tract of stranded green turtles, Chelonia mydas[J]. AIMS Microbiology, 2020, 6(4): 361-378. doi: 10.3934/microbiol.2020022







 DownLoad:
DownLoad: