Citation: Inês Barros, Hugo Froufe, George Marnellos, Conceição Egas, Jennifer Delaney, Michele Clamp, Ricardo Serrão Santos, Raul Bettencourt. Metatranscriptomics profile of the gill microbial community during Bathymodiolus azoricus aquarium acclimatization at atmospheric pressure[J]. AIMS Microbiology, 2018, 4(2): 240-260. doi: 10.3934/microbiol.2018.2.240

| [1] |
Wang H, Zhang H, Wong YH, et al. (2010) Rapid transcriptome and proteome profiling of a non-model marine invertebrate, Bugula neritina. Proteomics 10: 2972–2981. doi: 10.1002/pmic.201000056

|
| [2] | Childress J, Fisher C (1992) The biology of hydrothermal vent animals-physiology, biochemistry, and autotrophic symbioses. Oceanogr Mar Biol 30: 337–441. |
| [3] | Cavanaugh C, McKiness Z, Newton I, et al. (2005) Marine Chemosynthetic Symbioses, In: The Prokaryotes, 3 Eds. |
| [4] | Duperron S, Bergin C, Zielinski F, et al. (2006) A dual symbiosis shared by two mussel species, Bathymodiolus azoricus and B. puteoserpentis (Bivalvia: Mytilidae) from hydrothermal vents along the Mid-Atlantic Ridge. Environ Microbiol 8: 1441–1447. |
| [5] |
Egas C, Pinheiro M, Gomes P, et al. (2012) The transcriptome of Bathymodiolus azoricus gill reveals expression of genes from endosymbionts and free-living deep-sea bacteria. Mar Drugs 10: 1765–1783. doi: 10.3390/md10081765
|
| [6] |
Dubilier N, Bergin C, Lott C (2008) Symbiotic diversity in marine animals: the art of harnessing chemosynthesis. Nat Rev Microbiol 6: 725–740. doi: 10.1038/nrmicro1992
|
| [7] |
Canesi L, Gallo G, Gavioli M, et al. (2002) Bacteria-hemocyte interactions and phagocytosis in marine bivalves. Microsc Res Techniq 57: 469–476. doi: 10.1002/jemt.10100
|
| [8] |
Bettencourt R, Rodrigues M, Barros I, et al. (2014) Site-related differences in gene expression and bacterial densities in the mussel Bathymodiolus azoricus from the Menez Gwen and Lucky Strike deep-sea hydrothermal vent sites. Fish Shellfish Immun 39: 343–353. doi: 10.1016/j.fsi.2014.05.024
|
| [9] |
Bettencourt R, Pinheiro M, Egas C, et al. (2010) High-throughput sequencing and analysis of the gill tissue transcriptome from the deep-sea hydrothermal vent mussel Bathymodiolus azoricus. BMC Genomics 11: 559. doi: 10.1186/1471-2164-11-559
|
| [10] |
Franzosa EA, Hsu T, Sirota-Madi A, et al. (2015) Sequencing and beyond: integrating molecular "omics" for microbial community profiling. Nat Rev Microbiol 13: 360–372. doi: 10.1038/nrmicro3451
|
| [11] |
Turner TR, Ramakrishnan K, Walshaw J, et al. (2013) Comparative metatranscriptomics reveals kingdom level changes in the rhizosphere microbiome of plants. ISME J 7: 2248–2258. doi: 10.1038/ismej.2013.119
|
| [12] | Stewart FJ, Dmytrenko O, Delong EF, et al. (2011) Metatranscriptomic analysis of sulfur oxidation genes in the endosymbiont of Solemya velum. Front Microbiol 2: 134. |
| [13] |
Suárez-Ulloa V, Fernández-Tajes J, Manfrin C, et al. (2013) Bivalve omics: state of the art and potential applications for the biomonitoring of harmful marine compounds. Mar Drugs 11: 4370–4389. doi: 10.3390/md11114370
|
| [14] |
Gómez-Chiarri M, Guo X, Tanguy A, et al. (2015) The use of -omic tools in the study of disease processes in marine bivalve mollusks. J Invertebr Pathol 131: 137–154. doi: 10.1016/j.jip.2015.05.007
|
| [15] | Martin M (2011) Cutadapt removes adapter sequences from high-throughput sequencing reads. Embnet J 17: 10–12. |
| [16] |
Schmieder R, Edwards R (2011) Quality control and preprocessing of metagenomic datasets. Bioinformatics 27: 863–864. doi: 10.1093/bioinformatics/btr026
|
| [17] |
Kopylova E, Noé L, Touzet H (2012) SortMeRNA: fast and accurate filtering of ribosomal RNAs in metatranscriptomic data. Bioinformatics 28: 3211–3217. doi: 10.1093/bioinformatics/bts611
|
| [18] |
Caporaso JG, Kuczynski J, Stombaugh J, et al. (2010) QIIME allows analysis of high-throughput community sequencing data. Nat Methods 7: 335–336. doi: 10.1038/nmeth.f.303
|
| [19] |
Sinclair L, Osman OA, Bertilsson S, et al. (2015) Microbial community composition and diversity via 16s rrna gene amplicons: evaluating the illumina platform. PLoS One 10: e0116955. doi: 10.1371/journal.pone.0116955
|
| [20] |
Zielinski FU, Pernthaler A, Duperron S, et al. (2009) Widespread occurrence of an intranuclear bacterial parasite in vent and seep bathymodiolin mussels. Environ Microbiol 11: 1150–1167. doi: 10.1111/j.1462-2920.2008.01847.x
|
| [21] |
Jensen S, Duperron S, Birkeland NK, et al. (2010) Intracellular Oceanospirillales bacteria inhabit gills of Acesta bivalves. FEMS Microbiol Ecol 74: 523–533. doi: 10.1111/j.1574-6941.2010.00981.x
|
| [22] |
Beinart RA, Nyholm SV, Dubilier N, et al. (2014) Intracellular Oceanospirillales inhabit the gills of the hydrothermal vent snail Alviniconcha with chemosynthetic, γ-Proteobacterial symbionts. Environ Microbiol Rep 6: 656–664. doi: 10.1111/1758-2229.12183
|
| [23] |
Crépeau V, Cambon Bonavita MA, Lesongeur F, et al. (2011) Diversity and function in microbial mats from the Lucky Strike hydrothermal vent field. FEMS Microbiol Ecol 76: 524–540. doi: 10.1111/j.1574-6941.2011.01070.x
|
| [24] | Barros I, Divya B, Martins I, et al. (2014) Post-capture immune gene expression studies in the deep-sea hydrothermal vent mussel Bathymodiolus azoricus acclimatized to atmospheric pressure. Fish Shellfish Immun 42: 159–170. |
| [25] |
Fiala-Medioni A, McKiness Z, Dando P, et al. (2002) Ultrastructural, biochemical, and immunological characterization of two populations of the mytilid mussel Bathymodiolus azoricus from the Mid-Atlantic Ridge: evidence for a dual symbiosis. Mar Biol 141: 1035–1043. doi: 10.1007/s00227-002-0903-9
|
| [26] |
Szafranski KM, Piquet B, Shillito B, et al. (2015) Relative abundances of methane- and sulfur-oxidizing symbionts in gills of the deep-sea hydrothermal vent mussel Bathymodiolus azoricus under pressure. Deep-Sea Res Pt I 101: 7–13. doi: 10.1016/j.dsr.2015.03.003
|
| [27] |
Kádár E, Bettencourt R, Costa V, et al. (2005) Experimentally induced endosymbiont loss and re-acquirement in the hydrothermal vent bivalve Bathymodiolus azoricus. J Exp Mar Biol Ecol 318: 99–110. doi: 10.1016/j.jembe.2004.12.025
|
| [28] |
Pruski A, Rousse N, Fiala-Medioni A, et al. (2002) Sulphur signature in the hydrothermal vent mussel Bathymodiolus azoricus from the Mid-Atlantic Ridge. J Mar Biol Assoc UK 82: 463–468. doi: 10.1017/S0025315402005726
|
| [29] | Lodish H, Berk A, Zipursky SL, et al. (2000) Oxidation of Glucose and Fatty Acids to CO2, In: Freeman WH, Editor, Molecular Cell Biology, 4 Eds., New York. |
| [30] |
Taylor SS, Kornev AP (2011) Protein kinases: evolution of dynamic regulatory proteins. Trends Biochem Sci 36: 65–77. doi: 10.1016/j.tibs.2010.09.006
|
| [31] | Olmedo P, Hernandez AF, Pla A, et al. (2013) Determination of essential elements (copper, manganese, selenium and zinc) in fish and shellfish samples. Risk and nutritional assessment and mercury-selenium balance. Food Chem Toxicol 62: 299–307. |
| [32] |
Boutet I, Ripp R, Lecompte O, et al. (2011) Conjugating effects of symbionts and environmental factors on gene expression in deep-sea hydrothermal vent mussels. BMC Genomics 12: 530. doi: 10.1186/1471-2164-12-530
|
| [33] |
Varotto L, Domeneghetti S, Rosani U, et al. (2013) DNA damage and transcriptional changes in the gills of Mytilus galloprovincialis exposed to nanomolar doses of combined metal salts (Cd, Cu, Hg). PLoS One 8: e54602. doi: 10.1371/journal.pone.0054602
|
| [34] |
Tomanek L, Zuzow MJ, Hitt L, et al. (2012) Proteomics of hyposaline stress in blue mussel congeners (genus Mytilus): implications for biogeographic range limits in response to climate change. J Exp Biol 215: 3905–3916. doi: 10.1242/jeb.076448
|
| [35] |
Feder ME, Hofmann GE (1999) Heat-shock proteins, molecular chaperones, and the stress response: evolutionary and ecological physiology. Annu Rev Physiol 61: 243–282. doi: 10.1146/annurev.physiol.61.1.243
|
| [36] |
Pruski AM, Dixon DR (2007) Heat shock protein expression pattern (HSP70) in the hydrothermal vent mussel Bathymodiolus azoricus. Mar Environ Res 64: 209–224. doi: 10.1016/j.marenvres.2007.01.003
|
| [37] |
Bayne CJ (1990) Phagocytosis and non-self recognition in invertebrates. Bioscience 40: 723–731. doi: 10.2307/1311504
|
| [38] |
Bettencourt R, Dando P, Collins P, et al. (2009) Innate immunity in the deep sea hydrothermal vent mussel Bathymodiolus azoricus. Comp Biochem Phys A 152: 278–289. doi: 10.1016/j.cbpa.2008.10.022
|
| [39] | Bettencourt R, Barros I, Martins E, et al. (2017) An insightful model to study innate immunity and stress response in deep-sea vent animals: Profiling the mussel Bathymodiolus azoricus, In: Saja R, Editor, Organismal and Molecular Malacology, InTech. |
 microbiol-04-02-240-s1.pdf microbiol-04-02-240-s1.pdf |
 |
| microbiol-04-02-240-s2.pdf |
|
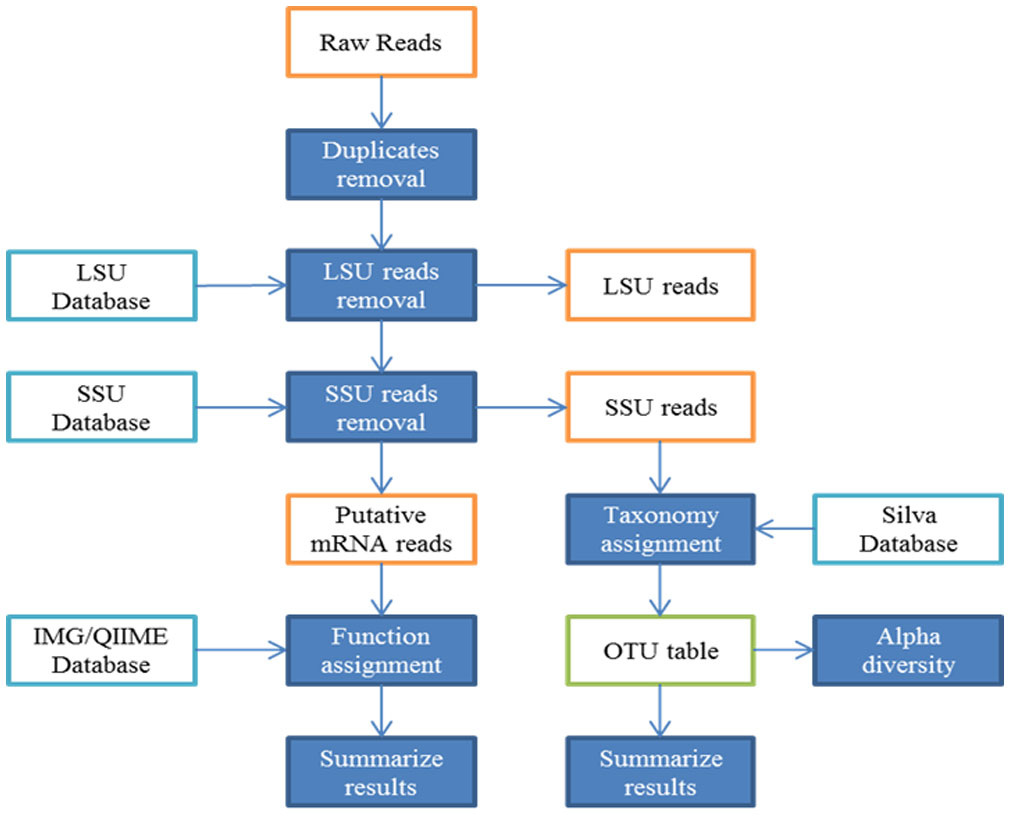
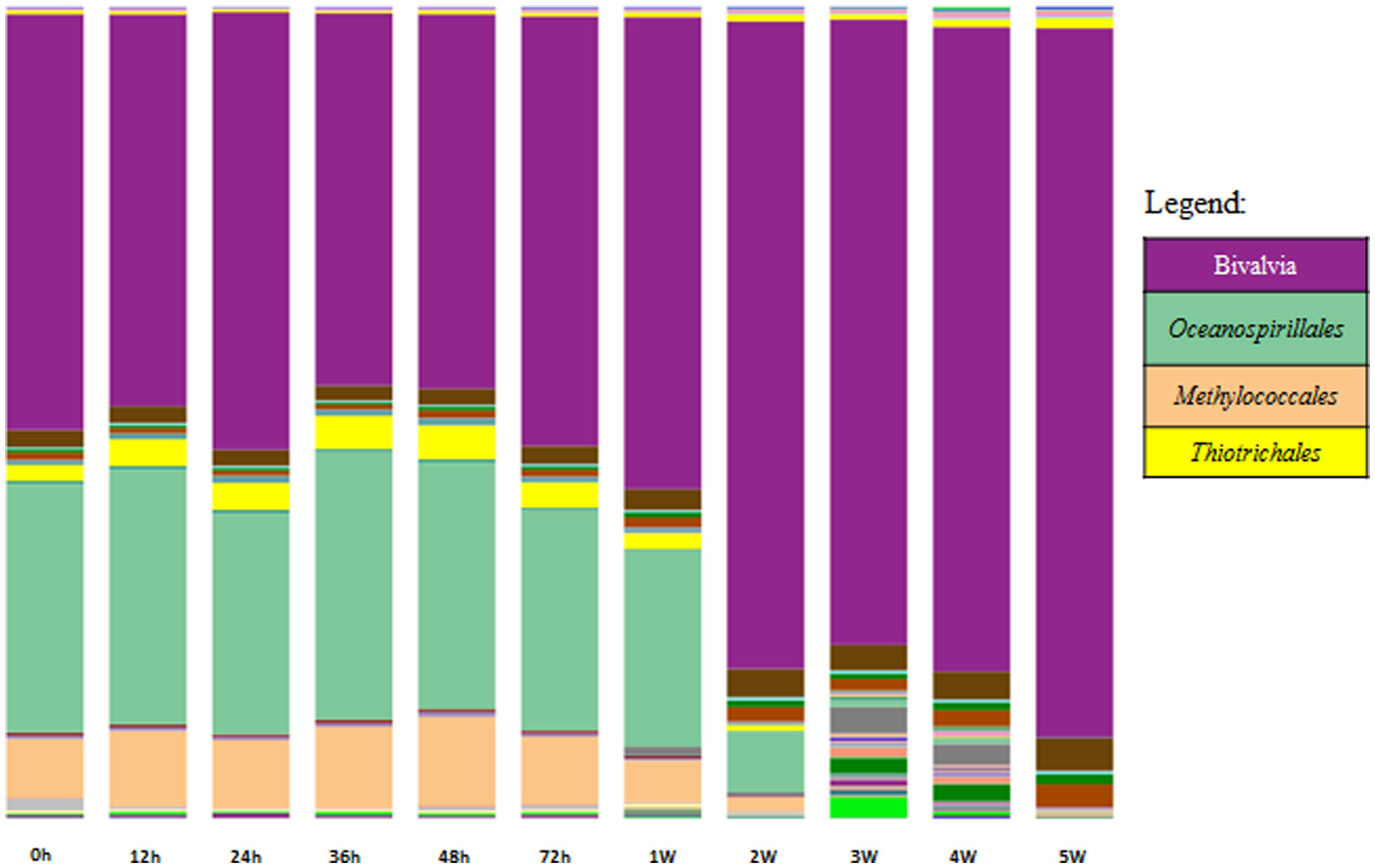
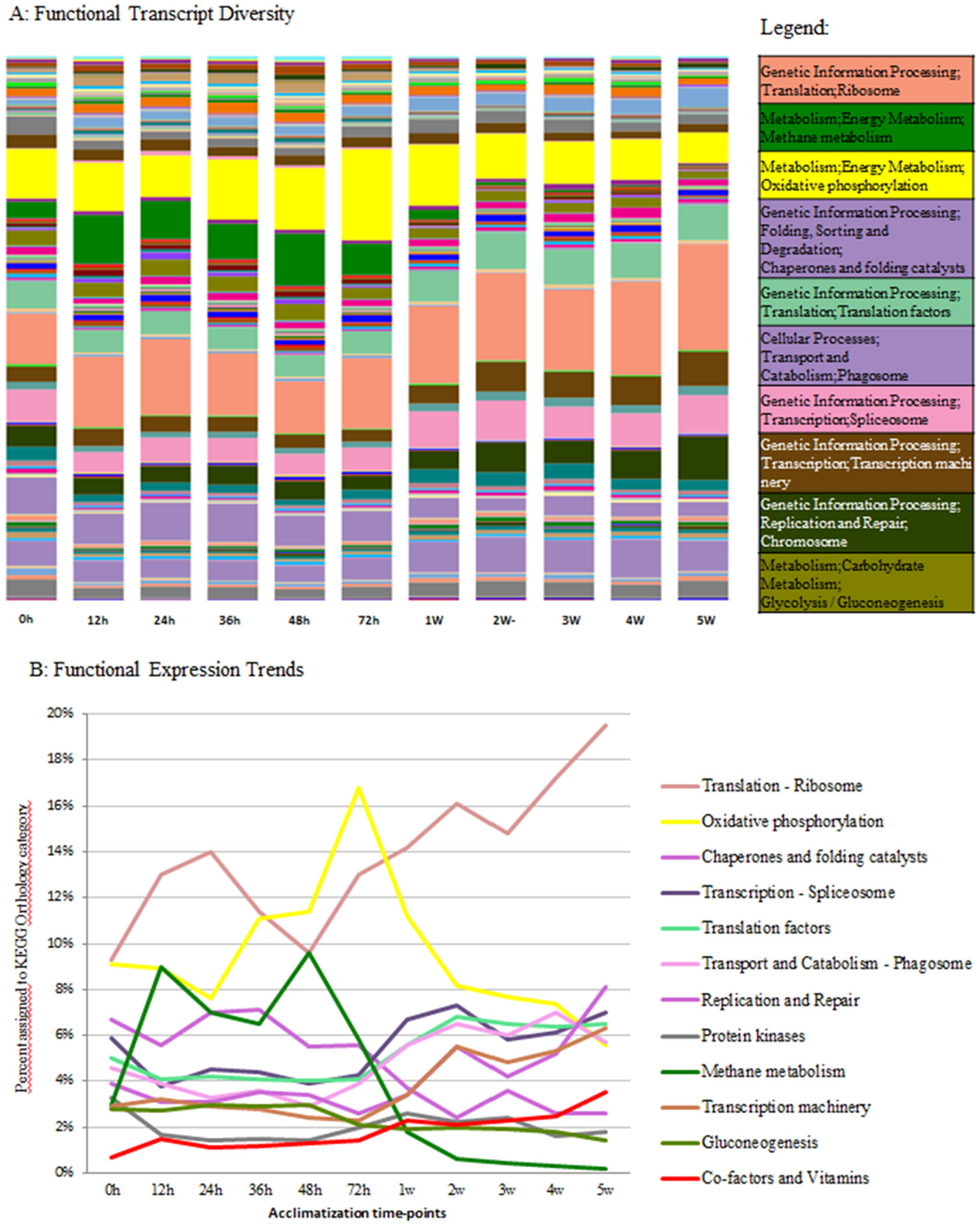
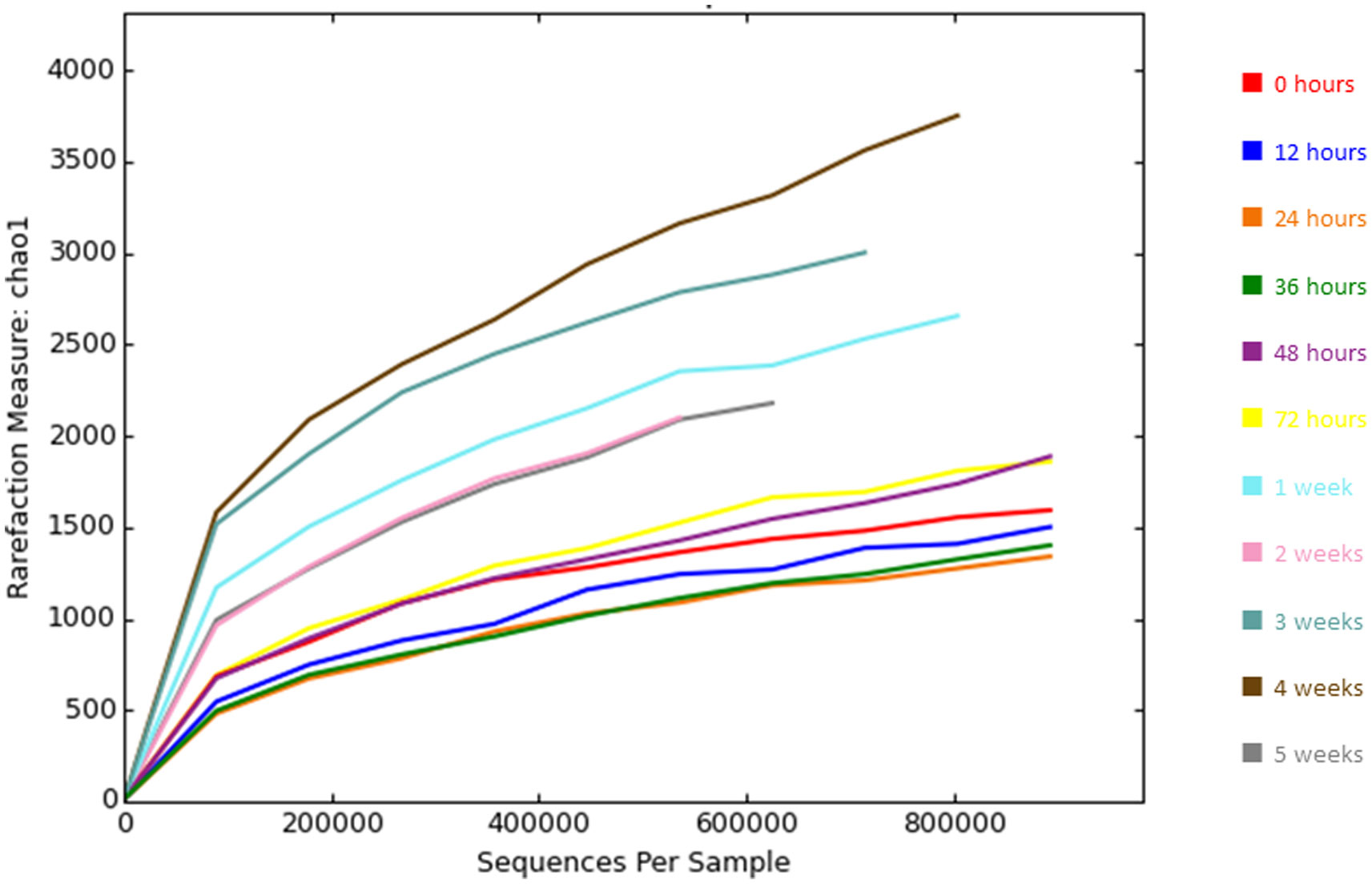
Figures(4) / Tables(2)

Inês Barros, Hugo Froufe, George Marnellos, Conceição Egas, Jennifer Delaney, Michele Clamp, Ricardo Serrão Santos, Raul Bettencourt. Metatranscriptomics profile of the gill microbial community during Bathymodiolus azoricus aquarium acclimatization at atmospheric pressure[J]. AIMS Microbiology, 2018, 4(2): 240-260. doi: 10.3934/microbiol.2018.2.240







 DownLoad:
DownLoad: