Citation: Michael J. Hamilton, Matthew D. Young, Silvia Sauer, Ernest Martinez. The interplay of long non-coding RNAs and MYC in cancer[J]. AIMS Biophysics, 2015, 2(4): 794-809. doi: 10.3934/biophy.2015.4.794

| [1] |
Volders PJ, Verheggen K, Menschaert G, et al. (2015) An update on LNCipedia: a database for annotated human lncRNA sequences. Nucleic Acids Res 43: D174-180. doi: 10.1093/nar/gku1060

|
| [2] | Amati B, Frank SR, Donjerkovic D, et al. (2001) Function of the c-Myc oncoprotein in chromatin remodeling and transcription. Biochim Biophys Acta 1471: M135-145. |
| [3] |
Bretones G, Delgado MD, Leon J (2015) Myc and cell cycle control. Biochim Biophys Acta 1849: 506-516. doi: 10.1016/j.bbagrm.2014.03.013
|
| [4] | Dang CV (2013) MYC, metabolism, cell growth, and tumorigenesis. Cold Spring Harb Perspect Med 3. |
| [5] |
McMahon SB (2014) MYC and the control of apoptosis. Cold Spring Harb Perspect Med 4: a014407. doi: 10.1101/cshperspect.a014407
|
| [6] | Vennstrom B, Sheiness D, Zabielski J, et al. (1982) Isolation and characterization of c-myc, a cellular homolog of the oncogene (v-myc) of avian myelocytomatosis virus strain 29. J Virol 42: 773-779. |
| [7] |
Zheng GX, Do BT, Webster DE, et al. (2014) Dicer-microRNA-Myc circuit promotes transcription of hundreds of long noncoding RNAs. Nat Struct Mol Biol 21: 585-590. doi: 10.1038/nsmb.2842
|
| [8] |
Winkle M, van den Berg A, Tayari M, et al. (2015) Long noncoding RNAs as a novel component of the Myc transcriptional network. FASEB J 29: 2338-2346. doi: 10.1096/fj.14-263889
|
| [9] |
Xiang JF, Yang L, Chen LL (2015) The long noncoding RNA regulation at the MYC locus. Curr Opin Genet Dev 33: 41-48. doi: 10.1016/j.gde.2015.07.001
|
| [10] |
Rinn JL, Chang HY (2012) Genome regulation by long noncoding RNAs. Annu Rev Biochem 81: 145-166. doi: 10.1146/annurev-biochem-051410-092902
|
| [11] |
Kapranov P, Cawley SE, Drenkow J, et al. (2002) Large-scale transcriptional activity in chromosomes 21 and 22. Science 296: 916-919. doi: 10.1126/science.1068597
|
| [12] |
Rinn JL, Euskirchen G, Bertone P, et al. (2003) The transcriptional activity of human Chromosome 22. Genes Dev 17: 529-540. doi: 10.1101/gad.1055203
|
| [13] |
Guttman M, Garber M, Levin JZ, et al. (2010) Ab initio reconstruction of cell type-specific transcriptomes in mouse reveals the conserved multi-exonic structure of lincRNAs. Nat Biotechnol 28: 503-510. doi: 10.1038/nbt.1633
|
| [14] |
Cabili MN, Trapnell C, Goff L, et al. (2011) Integrative annotation of human large intergenic noncoding RNAs reveals global properties and specific subclasses. Genes Dev 25: 1915-1927. doi: 10.1101/gad.17446611
|
| [15] |
Wu J, Okada T, Fukushima T, et al. (2012) A novel hypoxic stress-responsive long non-coding RNA transcribed by RNA polymerase III in Arabidopsis. RNA Biol 9: 302-313. doi: 10.4161/rna.19101
|
| [16] |
Derrien T, Johnson R, Bussotti G, et al. (2012) The GENCODE v7 catalog of human long noncoding RNAs: analysis of their gene structure, evolution, and expression. Genome Res 22: 1775-1789. doi: 10.1101/gr.132159.111
|
| [17] |
Gardini A, Shiekhattar R (2015) The many faces of long noncoding RNAs. FEBS J 282: 1647-1657. doi: 10.1111/febs.13101
|
| [18] |
Wilusz JE, Spector DL (2010) An unexpected ending: noncanonical 3' end processing mechanisms. RNA 16: 259-266. doi: 10.1261/rna.1907510
|
| [19] |
Zhang Y, Yang L, Chen LL (2014) Life without A tail: new formats of long noncoding RNAs. Int J Biochem Cell Biol 54: 338-349. doi: 10.1016/j.biocel.2013.10.009
|
| [20] |
Peart N, Sataluri A, Baillat D, et al. (2013) Non-mRNA 3' end formation: how the other half lives. Wiley Interdiscip Rev RNA 4: 491-506. doi: 10.1002/wrna.1174
|
| [21] | Ravasi T, Suzuki H, Pang KC, et al. (2006) Experimental validation of the regulated expression of large numbers of non-coding RNAs from the mouse genome. Genome Res 16: 11-19. |
| [22] |
Djebali S, Davis CA, Merkel A, et al. (2012) Landscape of transcription in human cells. Nature 489: 101-108. doi: 10.1038/nature11233
|
| [23] |
He S, Liu S, Zhu H (2011) The sequence, structure and evolutionary features of HOTAIR in mammals. BMC Evol Biol 11: 102. doi: 10.1186/1471-2148-11-102
|
| [24] |
Brown JA, Bulkley D, Wang J, et al. (2014) Structural insights into the stabilization of MALAT1 noncoding RNA by a bipartite triple helix. Nat Struct Mol Biol 21: 633-640. doi: 10.1038/nsmb.2844
|
| [25] |
Brown JA, Valenstein ML, Yario TA, et al. (2012) Formation of triple-helical structures by the 3'-end sequences of MALAT1 and MENbeta noncoding RNAs. Proc Natl Acad Sci U S A 109: 19202-19207. doi: 10.1073/pnas.1217338109
|
| [26] |
Smith MA, Gesell T, Stadler PF, et al. (2013) Widespread purifying selection on RNA structure in mammals. Nucleic Acids Res 41: 8220-8236. doi: 10.1093/nar/gkt596
|
| [27] |
Somarowthu S, Legiewicz M, Chillon I, et al. (2015) HOTAIR forms an intricate and modular secondary structure. Mol Cell 58: 353-361. doi: 10.1016/j.molcel.2015.03.006
|
| [28] |
Mortimer SA, Kidwell MA, Doudna JA (2014) Insights into RNA structure and function from genome-wide studies. Nat Rev Genet 15: 469-479. doi: 10.1038/nrg3681
|
| [29] | Ding Y, Tang Y, Kwok CK, et al. (2014) In vivo genome-wide profiling of RNA secondary structure reveals novel regulatory features. Nature 505: 696-700. |
| [30] |
Kertesz M, Wan Y, Mazor E, et al. (2010) Genome-wide measurement of RNA secondary structure in yeast. Nature 467: 103-107. doi: 10.1038/nature09322
|
| [31] |
Lucks JB, Mortimer SA, Trapnell C, et al. (2011) Multiplexed RNA structure characterization with selective 2'-hydroxyl acylation analyzed by primer extension sequencing (SHAPE-Seq). Proc Natl Acad Sci U S A 108: 11063-11068. doi: 10.1073/pnas.1106501108
|
| [32] |
Seetin MG, Kladwang W, Bida JP, et al. (2014) Massively parallel RNA chemical mapping with a reduced bias MAP-seq protocol. Methods Mol Biol 1086: 95-117. doi: 10.1007/978-1-62703-667-2_6
|
| [33] |
Underwood JG, Uzilov AV, Katzman S, et al. (2010) FragSeq: transcriptome-wide RNA structure probing using high-throughput sequencing. Nat Methods 7: 995-1001. doi: 10.1038/nmeth.1529
|
| [34] |
Aviran S, Trapnell C, Lucks JB, et al. (2011) Modeling and automation of sequencing-based characterization of RNA structure. Proc Natl Acad Sci U S A 108: 11069-11074. doi: 10.1073/pnas.1106541108
|
| [35] | Rouskin S, Zubradt M, Washietl S, et al. (2014) Genome-wide probing of RNA structure reveals active unfolding of mRNA structures in vivo. Nature 505: 701-705. |
| [36] |
Novikova IV, Hennelly SP, Sanbonmatsu KY (2013) Tackling structures of long noncoding RNAs. Int J Mol Sci 14: 23672-23684. doi: 10.3390/ijms141223672
|
| [37] |
Fatica A, Bozzoni I (2014) Long non-coding RNAs: new players in cell differentiation and development. Nat Rev Genet 15: 7-21. doi: 10.1038/nri3777
|
| [38] |
Batista PJ, Chang HY (2013) Long noncoding RNAs: cellular address codes in development and disease. Cell 152: 1298-1307. doi: 10.1016/j.cell.2013.02.012
|
| [39] |
Batista PJ, Chang HY (2013) Cytotopic localization by long noncoding RNAs. Curr Opin Cell Biol 25: 195-199. doi: 10.1016/j.ceb.2012.12.001
|
| [40] |
Han X, Yang F, Cao H, et al. (2015) Malat1 regulates serum response factor through miR-133 as a competing endogenous RNA in myogenesis. FASEB J 29: 3054-3064. doi: 10.1096/fj.14-259952
|
| [41] | Yoon JH, Abdelmohsen K, Kim J, et al. (2013) Scaffold function of long non-coding RNA HOTAIR in protein ubiquitination. Nat Commun 4: 2939. |
| [42] |
McHugh CA, Chen CK, Chow A, et al. (2015) The Xist lncRNA interacts directly with SHARP to silence transcription through HDAC3. Nature 521: 232-236. doi: 10.1038/nature14443
|
| [43] |
Deng K, Guo X, Wang H, et al. (2014) The lncRNA-MYC regulatory network in cancer. Tumour Biol 35: 9497-9503. doi: 10.1007/s13277-014-2511-y
|
| [44] |
Beroukhim R, Mermel CH, Porter D, et al. (2010) The landscape of somatic copy-number alteration across human cancers. Nature 463: 899-905. doi: 10.1038/nature08822
|
| [45] | Colombo T, Farina L, Macino G, et al. (2015) PVT1: a rising star among oncogenic long noncoding RNAs. Biomed Res Int 2015: 304208. |
| [46] |
Walz S, Lorenzin F, Morton J, et al. (2014) Activation and repression by oncogenic MYC shape tumour-specific gene expression profiles. Nature 511: 483-487. doi: 10.1038/nature13473
|
| [47] |
Carramusa L, Contino F, Ferro A, et al. (2007) The PVT-1 oncogene is a Myc protein target that is overexpressed in transformed cells. J Cell Physiol 213: 511-518. doi: 10.1002/jcp.21133
|
| [48] | Tseng YY, Moriarity BS, Gong W, et al. (2014) PVT1 dependence in cancer with MYC copy-number increase. Nature 512: 82-86. |
| [49] |
Zanke BW, Greenwood CM, Rangrej J, et al. (2007) Genome-wide association scan identifies a colorectal cancer susceptibility locus on chromosome 8q24. Nat Genet 39: 989-994. doi: 10.1038/ng2089
|
| [50] |
Tenesa A, Farrington SM, Prendergast JG, et al. (2008) Genome-wide association scan identifies a colorectal cancer susceptibility locus on 11q23 and replicates risk loci at 8q24 and 18q21. Nat Genet 40: 631-637. doi: 10.1038/ng.133
|
| [51] |
Tomlinson I, Webb E, Carvajal-Carmona L, et al. (2007) A genome-wide association scan of tag SNPs identifies a susceptibility variant for colorectal cancer at 8q24.21. Nat Genet 39: 984-988. doi: 10.1038/ng2085
|
| [52] |
Nissan A, Stojadinovic A, Mitrani-Rosenbaum S, et al. (2012) Colon cancer associated transcript-1: a novel RNA expressed in malignant and pre-malignant human tissues. Int J Cancer 130: 1598-1606. doi: 10.1002/ijc.26170
|
| [53] |
Ahmadiyeh N, Pomerantz MM, Grisanzio C, et al. (2010) 8q24 prostate, breast, and colon cancer risk loci show tissue-specific long-range interaction with MYC. Proc Natl Acad Sci U S A 107: 9742-9746. doi: 10.1073/pnas.0910668107
|
| [54] |
Kim T, Cui R, Jeon YJ, et al. (2014) Long-range interaction and correlation between MYC enhancer and oncogenic long noncoding RNA CARLo-5. Proc Natl Acad Sci U S A 111: 4173-4178. doi: 10.1073/pnas.1400350111
|
| [55] |
Pomerantz MM, Ahmadiyeh N, Jia L, et al. (2009) The 8q24 cancer risk variant rs6983267 shows long-range interaction with MYC in colorectal cancer. Nat Genet 41: 882-884. doi: 10.1038/ng.403
|
| [56] |
Sur IK, Hallikas O, Vaharautio A, et al. (2012) Mice lacking a Myc enhancer that includes human SNP rs6983267 are resistant to intestinal tumors. Science 338: 1360-1363. doi: 10.1126/science.1228606
|
| [57] |
Tuupanen S, Turunen M, Lehtonen R, et al. (2009) The common colorectal cancer predisposition SNP rs6983267 at chromosome 8q24 confers potential to enhanced Wnt signaling. Nat Genet 41: 885-890. doi: 10.1038/ng.406
|
| [58] |
Xiang JF, Yin QF, Chen T, et al. (2014) Human colorectal cancer-specific CCAT1-L lncRNA regulates long-range chromatin interactions at the MYC locus. Cell Res 24: 513-531. doi: 10.1038/cr.2014.35
|
| [59] |
Deng L, Yang SB, Xu FF, et al. (2015) Long noncoding RNA CCAT1 promotes hepatocellular carcinoma progression by functioning as let-7 sponge. J Exp Clin Cancer Res 34: 18. doi: 10.1186/s13046-015-0136-7
|
| [60] |
He X, Tan X, Wang X, et al. (2014) C-Myc-activated long noncoding RNA CCAT1 promotes colon cancer cell proliferation and invasion. Tumour Biol 35: 12181-12188. doi: 10.1007/s13277-014-2526-4
|
| [61] |
Yang F, Xue X, Bi J, et al. (2013) Long noncoding RNA CCAT1, which could be activated by c-Myc, promotes the progression of gastric carcinoma. J Cancer Res Clin Oncol 139: 437-445. doi: 10.1007/s00432-012-1324-x
|
| [62] |
Ling H, Spizzo R, Atlasi Y, et al. (2013) CCAT2, a novel noncoding RNA mapping to 8q24, underlies metastatic progression and chromosomal instability in colon cancer. Genome Res 23: 1446-1461. doi: 10.1101/gr.152942.112
|
| [63] | Kim T, Jeon YJ, Cui R, et al. (2015) Role of MYC-regulated long noncoding RNAs in cell cycle regulation and tumorigenesis. J Natl Cancer Inst 107. |
| [64] | Ye Z, Zhou M, Tian B, et al. (2015) Expression of lncRNA-CCAT1, E-cadherin and N-cadherin in colorectal cancer and its clinical significance. Int J Clin Exp Med 8: 3707-3715. |
| [65] |
Alaiyan B, Ilyayev N, Stojadinovic A, et al. (2013) Differential expression of colon cancer associated transcript1 (CCAT1) along the colonic adenoma-carcinoma sequence. BMC Cancer 13: 196. doi: 10.1186/1471-2407-13-196
|
| [66] | Kim T, Cui R, Jeon YJ, et al. (2015) MYC-repressed long noncoding RNAs antagonize MYC-induced cell proliferation and cell cycle progression. Oncotarget. |
| [67] |
Prensner JR, Chen W, Han S, et al. (2014) The long non-coding RNA PCAT-1 promotes prostate cancer cell proliferation through cMyc. Neoplasia 16: 900-908. doi: 10.1016/j.neo.2014.09.001
|
| [68] |
Yamamura S, Saini S, Majid S, et al. (2012) MicroRNA-34a modulates c-Myc transcriptional complexes to suppress malignancy in human prostate cancer cells. PLoS One 7: e29722. doi: 10.1371/journal.pone.0029722
|
| [69] |
Siemens H, Jackstadt R, Hunten S, et al. (2011) miR-34 and SNAIL form a double-negative feedback loop to regulate epithelial-mesenchymal transitions. Cell Cycle 10: 4256-4271. doi: 10.4161/cc.10.24.18552
|
| [70] |
Benassi B, Flavin R, Marchionni L, et al. (2012) MYC is activated by USP2a-mediated modulation of microRNAs in prostate cancer. Cancer Discov 2: 236-247. doi: 10.1158/2159-8290.CD-11-0219
|
| [71] |
Poliseno L, Salmena L, Zhang J, et al. (2010) A coding-independent function of gene and pseudogene mRNAs regulates tumour biology. Nature 465: 1033-1038. doi: 10.1038/nature09144
|
| [72] |
Salmena L, Poliseno L, Tay Y, et al. (2011) A ceRNA hypothesis: the Rosetta Stone of a hidden RNA language? Cell 146: 353-358. doi: 10.1016/j.cell.2011.07.014
|
| [73] |
Ge X, Chen Y, Liao X, et al. (2013) Overexpression of long noncoding RNA PCAT-1 is a novel biomarker of poor prognosis in patients with colorectal cancer. Med Oncol 30: 588. doi: 10.1007/s12032-013-0588-6
|
| [74] |
Chung S, Nakagawa H, Uemura M, et al. (2011) Association of a novel long non-coding RNA in 8q24 with prostate cancer susceptibility. Cancer Sci 102: 245-252. doi: 10.1111/j.1349-7006.2010.01737.x
|
| [75] |
Hung CL, Wang LY, Yu YL, et al. (2014) A long noncoding RNA connects c-Myc to tumor metabolism. Proc Natl Acad Sci U S A 111: 18697-18702. doi: 10.1073/pnas.1415669112
|
| [76] |
Chu C, Qu K, Zhong FL, et al. (2011) Genomic maps of long noncoding RNA occupancy reveal principles of RNA-chromatin interactions. Mol Cell 44: 667-678. doi: 10.1016/j.molcel.2011.08.027
|
| [77] | Pickard MR, Williams GT (2015) Molecular and Cellular Mechanisms of Action of Tumour Suppressor GAS5 LncRNA. Genes (Basel) 6: 484-499. |
| [78] |
Hu G, Lou Z, Gupta M (2014) The long non-coding RNA GAS5 cooperates with the eukaryotic translation initiation factor 4E to regulate c-Myc translation. PLoS One 9: e107016. doi: 10.1371/journal.pone.0107016
|
| [79] |
Mourtada-Maarabouni M, Pickard MR, Hedge VL, et al. (2009) GAS5, a non-protein-coding RNA, controls apoptosis and is downregulated in breast cancer. Oncogene 28: 195-208. doi: 10.1038/onc.2008.373
|
| [80] |
Pickard MR, Mourtada-Maarabouni M, Williams GT (2013) Long non-coding RNA GAS5 regulates apoptosis in prostate cancer cell lines. Biochim Biophys Acta 1832: 1613-1623. doi: 10.1016/j.bbadis.2013.05.005
|
| [81] |
Sun M, Jin FY, Xia R, et al. (2014) Decreased expression of long noncoding RNA GAS5 indicates a poor prognosis and promotes cell proliferation in gastric cancer. BMC Cancer 14: 319. doi: 10.1186/1471-2407-14-319
|
| [82] | Tu ZQ, Li RJ, Mei JZ, et al. (2014) Down-regulation of long non-coding RNA GAS5 is associated with the prognosis of hepatocellular carcinoma. Int J Clin Exp Pathol 7: 4303-4309. |
| [83] | Li LJ, Zhu JL, Bao WS, et al. (2014) Long noncoding RNA GHET1 promotes the development of bladder cancer. Int J Clin Exp Pathol 7: 7196-7205. |
| [84] |
Yang F, Xue X, Zheng L, et al. (2014) Long non-coding RNA GHET1 promotes gastric carcinoma cell proliferation by increasing c-Myc mRNA stability. FEBS J 281: 802-813. doi: 10.1111/febs.12625
|
| [85] |
Lemm I, Ross J (2002) Regulation of c-myc mRNA decay by translational pausing in a coding region instability determinant. Mol Cell Biol 22: 3959-3969. doi: 10.1128/MCB.22.12.3959-3969.2002
|
| [86] | Weidensdorfer D, Stohr N, Baude A, et al. (2009) Control of c-myc mRNA stability by IGF2BP1-associated cytoplasmic RNPs. RNA 15: 104-115. |
| [87] |
Matouk IJ, DeGroot N, Mezan S, et al. (2007) The H19 non-coding RNA is essential for human tumor growth. PLoS One 2: e845. doi: 10.1371/journal.pone.0000845
|
| [88] |
Matouk IJ, Raveh E, Abu-lail R, et al. (2014) Oncofetal H19 RNA promotes tumor metastasis. Biochim Biophys Acta 1843: 1414-1426. doi: 10.1016/j.bbamcr.2014.03.023
|
| [89] | Jiang X, Yan Y, Hu M, et al. (2015) Increased level of H19 long noncoding RNA promotes invasion, angiogenesis, and stemness of glioblastoma cells. J Neurosurg: 1-8. |
| [90] |
Lottin S, Adriaenssens E, Dupressoir T, et al. (2002) Overexpression of an ectopic H19 gene enhances the tumorigenic properties of breast cancer cells. Carcinogenesis 23: 1885-1895. doi: 10.1093/carcin/23.11.1885
|
| [91] |
Luo M, Li Z, Wang W, et al. (2013) Long non-coding RNA H19 increases bladder cancer metastasis by associating with EZH2 and inhibiting E-cadherin expression. Cancer Lett 333: 213-221. doi: 10.1016/j.canlet.2013.01.033
|
| [92] |
Ma C, Nong K, Zhu H, et al. (2014) H19 promotes pancreatic cancer metastasis by derepressing let-7's suppression on its target HMGA2-mediated EMT. Tumour Biol 35: 9163-9169. doi: 10.1007/s13277-014-2185-5
|
| [93] |
Zhang EB, Han L, Yin DD, et al. (2014) c-Myc-induced, long, noncoding H19 affects cell proliferation and predicts a poor prognosis in patients with gastric cancer. Med Oncol 31: 914. doi: 10.1007/s12032-014-0914-7
|
| [94] |
Barsyte-Lovejoy D, Lau SK, Boutros PC, et al. (2006) The c-Myc oncogene directly induces the H19 noncoding RNA by allele-specific binding to potentiate tumorigenesis. Cancer Res 66: 5330-5337. doi: 10.1158/0008-5472.CAN-06-0037
|
| [95] |
Shi Y, Wang Y, Luan W, et al. (2014) Long non-coding RNA H19 promotes glioma cell invasion by deriving miR-675. PLoS One 9: e86295. doi: 10.1371/journal.pone.0086295
|
| [96] |
Kallen AN, Zhou XB, Xu J, et al. (2013) The imprinted H19 lncRNA antagonizes let-7 microRNAs. Mol Cell 52: 101-112. doi: 10.1016/j.molcel.2013.08.027
|
| [97] |
Yan L, Zhou J, Gao Y, et al. (2015) Regulation of tumor cell migration and invasion by the H19/let-7 axis is antagonized by metformin-induced DNA methylation. Oncogene 34: 3076-3084. doi: 10.1038/onc.2014.236
|
| [98] |
Liao LM, Sun XY, Liu AW, et al. (2014) Low expression of long noncoding XLOC_010588 indicates a poor prognosis and promotes proliferation through upregulation of c-Myc in cervical cancer. Gynecol Oncol 133: 616-623. doi: 10.1016/j.ygyno.2014.03.555
|
| [99] |
Mestdagh P, Fredlund E, Pattyn F, et al. (2010) An integrative genomics screen uncovers ncRNA T-UCR functions in neuroblastoma tumours. Oncogene 29: 3583-3592. doi: 10.1038/onc.2010.106
|
| [100] |
Atmadibrata B, Liu PY, Sokolowski N, et al. (2014) The novel long noncoding RNA linc00467 promotes cell survival but is down-regulated by N-Myc. PLoS One 9: e88112. doi: 10.1371/journal.pone.0088112
|
| [101] |
Tee AE, Ling D, Nelson C, et al. (2014) The histone demethylase JMJD1A induces cell migration and invasion by up-regulating the expression of the long noncoding RNA MALAT1. Oncotarget 5: 1793-1804. doi: 10.18632/oncotarget.1785
|
| [102] | Liu PY, Erriquez D, Marshall GM, et al. (2014) Effects of a novel long noncoding RNA, lncUSMycN, on N-Myc expression and neuroblastoma progression. J Natl Cancer Inst 106. |
| [103] |
Vadie N, Saayman S, Lenox A, et al. (2015) MYCNOS functions as an antisense RNA regulating MYCN. RNA Biol 12: 893-899. doi: 10.1080/15476286.2015.1063773
|
| [104] |
Stanton BR, Perkins AS, Tessarollo L, et al. (1992) Loss of N-myc function results in embryonic lethality and failure of the epithelial component of the embryo to develop. Genes Dev 6: 2235-2247. doi: 10.1101/gad.6.12a.2235
|
| [105] | Stanton BR, Parada LF (1992) The N-myc proto-oncogene: developmental expression and in vivo site-directed mutagenesis. Brain Pathol 2: 71-83. |
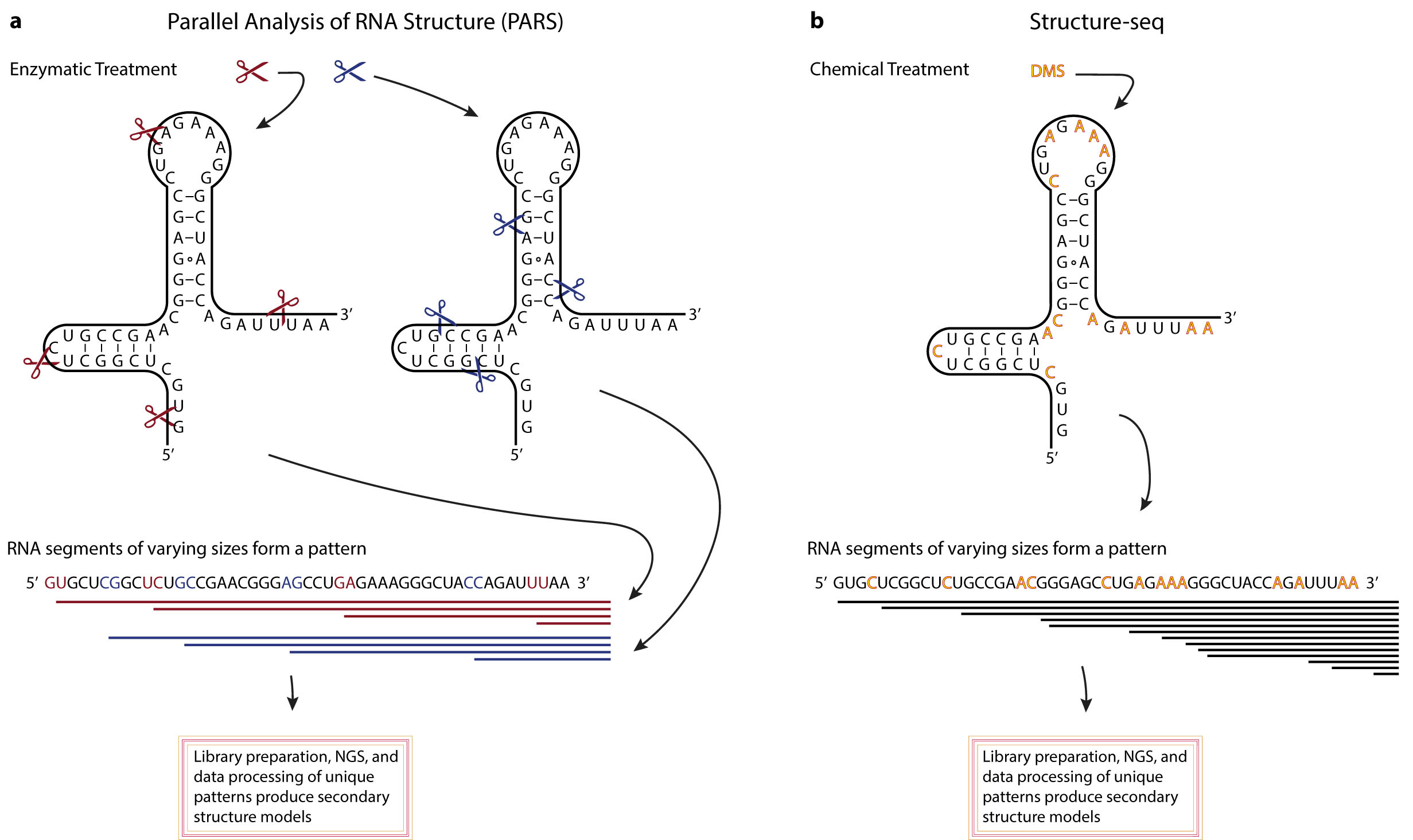
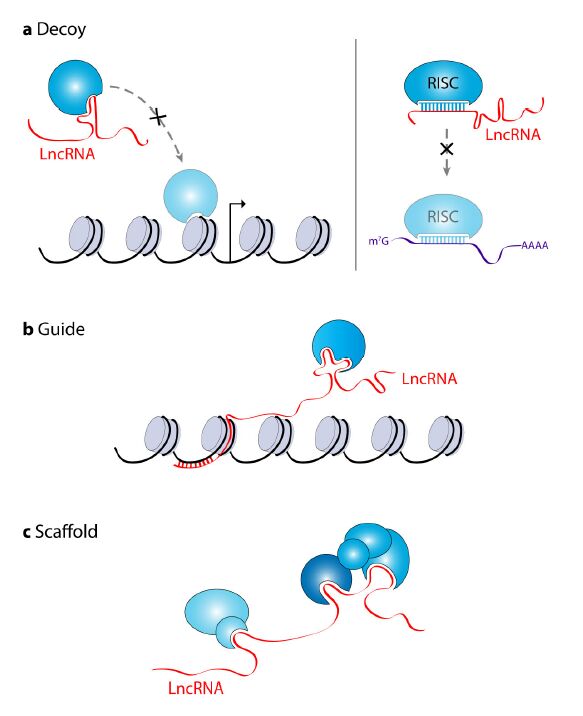
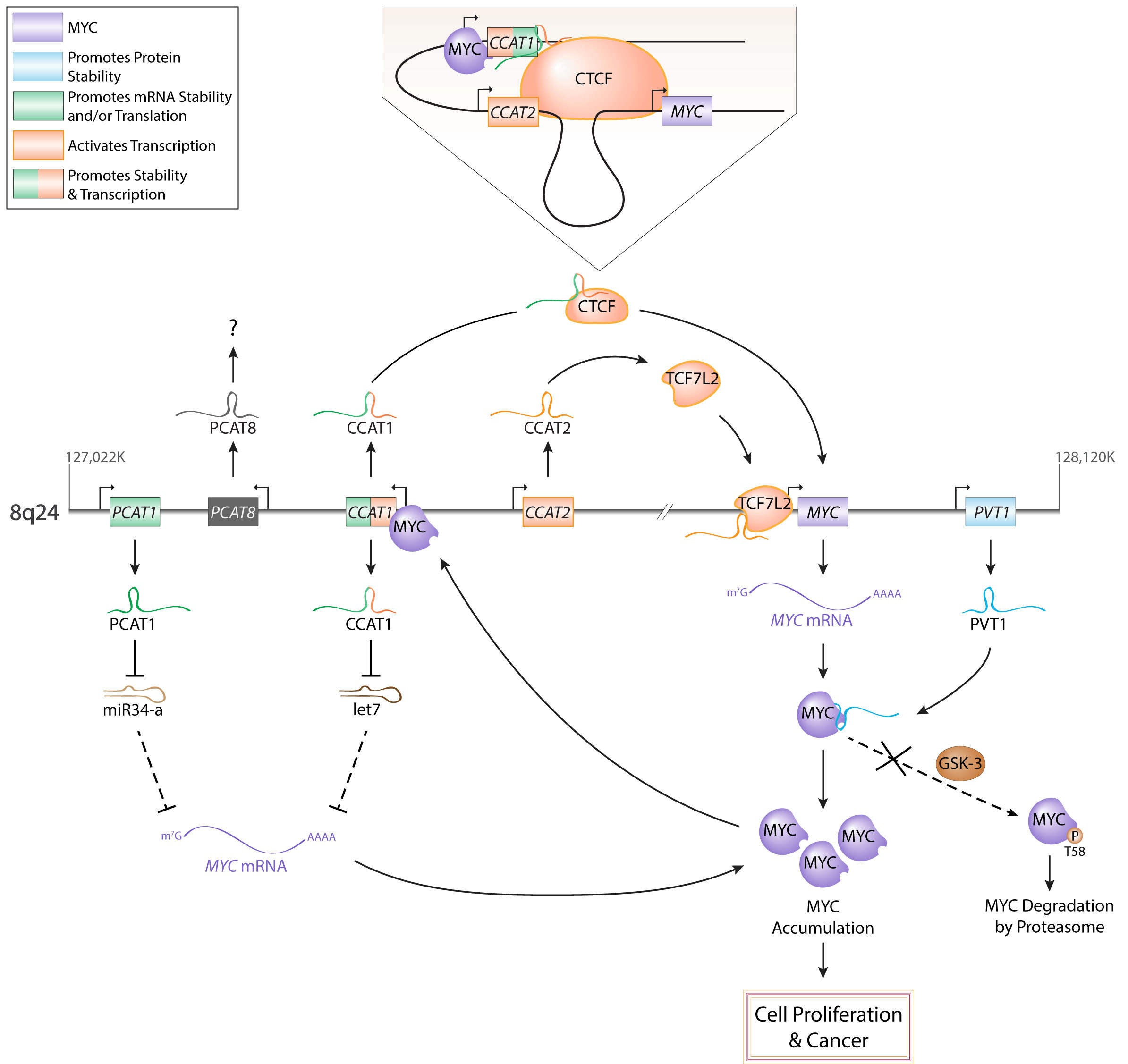
Figures(3) / Tables(1)

Michael J. Hamilton, Matthew D. Young, Silvia Sauer, Ernest Martinez. The interplay of long non-coding RNAs and MYC in cancer[J]. AIMS Biophysics, 2015, 2(4): 794-809. doi: 10.3934/biophy.2015.4.794







 DownLoad:
DownLoad: