Citation: Laetitia Weinhard, Paolo d'Errico, Tuan Leng Tay. Headmasters: Microglial regulation of learning and memory in health and disease[J]. AIMS Molecular Science, 2018, 5(1): 63-89. doi: 10.3934/molsci.2018.1.63

| [1] |
Alliot F, Godin I, Pessac B (1999) Microglia derive from progenitors, originating from the yolk sac, and which proliferate in the brain. Brain Res Dev Brain Res 117: 145–152. doi: 10.1016/S0165-3806(99)00113-3

|
| [2] |
Ginhoux F, Greter M, Leboeuf M, et al. (2010) Fate mapping analysis reveals that adult microglia derive from primitive macrophages. Science 330: 841–845. doi: 10.1126/science.1194637
|
| [3] | Gomez Perdiguero E, Klapproth K, Schulz C, et al. (2015) Tissue-resident macrophages originate from yolk-sac-derived erythro-myeloid progenitors. Nature 518: 547–551. |
| [4] |
Tay TL, Hagemeyer N, Prinz M (2016) The force awakens: Insights into the origin and formation of microglia. Curr Opin Neurobiol 39: 30–37. doi: 10.1016/j.conb.2016.04.003
|
| [5] |
Tay TL, Savage JC, Hui CW, et al. (2017) Microglia across the lifespan: From origin to function in brain development, plasticity and cognition. J Physiol 595: 1929–1945. doi: 10.1113/JP272134
|
| [6] |
Askew K, Li K, Olmos-Alonso A, et al. (2017) Coupled proliferation and apoptosis maintain the rapid turnover of microglia in the adult brain. Cell Rep 18: 391–405. doi: 10.1016/j.celrep.2016.12.041
|
| [7] |
Lawson LJ, Perry VH, Gordon S (1992) Turnover of resident microglia in the normal adult mouse brain. Neuroscience 48: 405–415. doi: 10.1016/0306-4522(92)90500-2
|
| [8] |
Réu P, Khosravi A, Bernard S, et al. (2017) The lifespan and turnover of microglia in the human brain. Cell Rep 20: 779–784. doi: 10.1016/j.celrep.2017.07.004
|
| [9] |
Tay TL, Mai D, Dautzenberg J, et al. (2017) A new fate mapping system reveals context-dependent random or clonal expansion of microglia. Nat Neurosci 20: 793–803. doi: 10.1038/nn.4547
|
| [10] |
Davalos D, Grutzendler J, Yang G, et al. (2005) ATP mediates rapid microglial response to local brain injury in vivo. Nat Neurosci 8: 752–758. doi: 10.1038/nn1472
|
| [11] |
Nimmerjahn A, Kirchhoff F, Helmchen F (2005) Resting microglial cells are highly dynamic surveillants of brain parenchyma in vivo. Science 308: 1314–1318. doi: 10.1126/science.1110647
|
| [12] |
Kettenmann H, Hanisch UK, Noda M, et al. (2011) Physiology of microglia. Physiol Rev 91: 461. doi: 10.1152/physrev.00011.2010
|
| [13] |
Arnoux I, Hoshiko M, Mandavy L, et al. (2013) Adaptive phenotype of microglial cells during the normal postnatal development of the somatosensory "barrel" cortex. Glia 61: 1582–1594. doi: 10.1002/glia.22503
|
| [14] |
Clark AK, Gruber-Schoffnegger D, Drdla-Schutting R, et al. (2015) Selective activation of microglia facilitates synaptic strength. J Neurosci 35: 4552–4570. doi: 10.1523/JNEUROSCI.2061-14.2015
|
| [15] |
Li Y, Du XF, Liu CS, et al. (2012) Reciprocal regulation between resting microglial dynamics and neuronal activity in vivo. Dev Cell 23: 1189–1202. doi: 10.1016/j.devcel.2012.10.027
|
| [16] |
Schafer DP, Lehrman EK, Kautzman AG, et al. (2012) Microglia sculpt postnatal neural circuits in an activity and complement-dependent manner. Neuron 74: 691–705. doi: 10.1016/j.neuron.2012.03.026
|
| [17] |
Arnò B, Grassivaro F, Rossi C, et al. (2014) Neural progenitor cells orchestrate microglia migration and positioning into the developing cortex. Nat Commun 5: 5611. doi: 10.1038/ncomms6611
|
| [18] |
Hagemeyer N, Hanft KM, Akriditou MA, et al. (2017) Microglia contribute to normal myelinogenesis and to oligodendrocyte progenitor maintenance during adulthood. Acta Neuropathol 134: 441–458. doi: 10.1007/s00401-017-1747-1
|
| [19] |
Shigemoto-Mogami Y, Hoshikawa K, Goldman JE, et al. (2014) Microglia enhance neurogenesis and oligodendrogenesis in the early postnatal subventricular zone. J Neurosci 34: 2231–2243. doi: 10.1523/JNEUROSCI.1619-13.2014
|
| [20] |
Ueno M, Fujita Y, Tanaka T, et al. (2013) Layer V cortical neurons require microglial support for survival during postnatal development. Nat Neurosci 16: 543–551. doi: 10.1038/nn.3358
|
| [21] |
Hoshiko M, Arnoux I, Avignone E, et al. (2012) Deficiency of the microglial receptor CX3CR1 impairs postnatal functional development of thalamocortical synapses in the barrel cortex. J Neurosci 32: 15106–15111. doi: 10.1523/JNEUROSCI.1167-12.2012
|
| [22] |
Paolicelli RC, Bolasco G, Pagani F, et al. (2011) Synaptic pruning by microglia is necessary for normal brain development. Science 333: 1456–1458. doi: 10.1126/science.1202529
|
| [23] |
Zhan Y, Paolicelli RC, Sforazzini F, et al. (2014) Deficient neuron-microglia signaling results in impaired functional brain connectivity and social behavior. Nat Neurosci 17: 400–406. doi: 10.1038/nn.3641
|
| [24] |
Cunningham CL, Martínez-Cerde?o V, Noctor SC (2013) Microglia regulate the number of neural precursor cells in the developing cerebral cortex. J Neurosci 33: 4216–4233. doi: 10.1523/JNEUROSCI.3441-12.2013
|
| [25] |
Marín-Teva JL, Dusart I, Colin C, et al. (2004) Microglia promote the death of developing Purkinje cells. Neuron 41: 535–547. doi: 10.1016/S0896-6273(04)00069-8
|
| [26] |
Peri F, Nüsslein-Volhard C (2008) Live imaging of neuronal degradation by microglia reveals a role for v0-ATPase a1 in phagosomal fusion in vivo. Cell 133: 916–927. doi: 10.1016/j.cell.2008.04.037
|
| [27] |
Swinnen N, Smolders S, Avila A, et al. (2013) Complex invasion pattern of the cerebral cortex by microglial cells during development of the mouse embryo. Glia 61: 150–163. doi: 10.1002/glia.22421
|
| [28] |
Brown GC, Neher JJ (2014) Microglial phagocytosis of live neurons. Nat Rev Neurosci 15: 209–216. doi: 10.1038/nrn3710
|
| [29] |
Ahlers KE, Kara?ay B, Fuller L, et al. (2015) Transient activation of microglia following acute alcohol exposure in developing mouse neocortex is primarily driven by BAX-dependent neurodegeneration. Glia 63: 1694–1713. doi: 10.1002/glia.22835
|
| [30] |
Anacker C, Hen R (2017) Adult hippocampal neurogenesis and cognitive flexibility - linking memory and mood. Nat Rev Neurosci 18: 335–346. doi: 10.1038/nrn.2017.45
|
| [31] | Sierra A, Beccari S, Diaz-Aparicio I, et al. (2014) Surveillance, phagocytosis, and inflammation: How never-resting microglia influence adult hippocampal neurogenesis. Neural Plast 2014: 610343. |
| [32] |
Sierra A, Encinas JM, Deudero JJP, et al. (2010) Microglia shape adult hippocampal neurogenesis through apoptosis-coupled phagocytosis. Cell Stem Cell 7: 483–495. doi: 10.1016/j.stem.2010.08.014
|
| [33] |
Ribeiro Xavier AL, Kress BT, Goldman SA, et al. (2015) A distinct population of microglia supports adult neurogenesis in the subventricular zone. J Neurosci 35: 11848–11861. doi: 10.1523/JNEUROSCI.1217-15.2015
|
| [34] |
Gu Y, Arruda-Carvalho M, Wang J, et al. (2012) Optical controlling reveals time-dependent roles for adult-born dentate granule cells. Nat Neurosci 15: 1700–1706. doi: 10.1038/nn.3260
|
| [35] |
Nakashiba T, Cushman JD, Pelkey KA, et al. (2012) Young dentate granule cells mediate pattern separation, whereas old granule cells facilitate pattern completion. Cell 149: 188–201. doi: 10.1016/j.cell.2012.01.046
|
| [36] |
Bachstetter AD, Morganti JM, Jernberg J, et al. (2011) Fractalkine and CXCR1 regulate hippocampal neurogenesis in adult and aged rats. Neurobiol Aging 32: 2030–2044. doi: 10.1016/j.neurobiolaging.2009.11.022
|
| [37] |
Rogers JT, Morganti JM, Bachstetter AD, et al. (2011) CX3CR1 deficiency leads to impairment of hippocampal cognitive function and synaptic plasticity. J Neurosci 31: 16241–16250. doi: 10.1523/JNEUROSCI.3667-11.2011
|
| [38] |
Sellner S, Paricio-Montesinos R, Spie? A, et al. (2016) Microglial CX3CR1 promotes adult neurogenesis by inhibiting Sirt 1/p65 signaling independent of CX3CL1. Acta Neuropathol Commun 4: 102. doi: 10.1186/s40478-016-0374-8
|
| [39] |
Citri A, Malenka RC (2008) Synaptic plasticity: Multiple forms, functions, and mechanisms. Neuropsychopharmacology 33: 18–41. doi: 10.1038/sj.npp.1301559
|
| [40] |
Neves G, Cooke SF, Bliss TVP (2008) Synaptic plasticity, memory and the hippocampus: A neural network approach to causality. Nat Rev Neurosci 9: 65–75. doi: 10.1038/nrn2303
|
| [41] |
Roumier A, Béchade C, Poncer JC, et al. (2004) Impaired synaptic function in the microglial KARAP/DAP12-deficient mouse. J Neurosci 24: 11421–11428. doi: 10.1523/JNEUROSCI.2251-04.2004
|
| [42] |
Roumier A, Pascual O, Béchade C, et al. (2008) Prenatal activation of microglia induces delayed impairment of glutamatergic synaptic function. PLoS One 3: e2595. doi: 10.1371/journal.pone.0002595
|
| [43] |
Costello DA, Lyons A, Denieffe S, et al. (2011) Long term potentiation is impaired in membrane glycoprotein CD200-deficient mice: A role for Toll-like receptor activation. J Biol Chem 286: 34722–34732. doi: 10.1074/jbc.M111.280826
|
| [44] |
Stellwagen D, Malenka RC (2006) Synaptic scaling mediated by glial TNF-alpha. Nature 440: 1054–1059. doi: 10.1038/nature04671
|
| [45] |
Santello M, Volterra A (2012) TNFα in synaptic function: Switching gears. Trends Neurosci 35: 638–647. doi: 10.1016/j.tins.2012.06.001
|
| [46] |
Arisi GM (2014) Nervous and immune systems signals and connections: Cytokines in hippocampus physiology and pathology. Epilepsy Behav 38: 43–47. doi: 10.1016/j.yebeh.2014.01.017
|
| [47] |
McAfoose J, Baune BT (2009) Evidence for a cytokine model of cognitive function. Neurosci Biobehav Rev 33: 355–366. doi: 10.1016/j.neubiorev.2008.10.005
|
| [48] |
Williamson LL, Sholar PW, Mistry RS, et al. (2011) Microglia and memory: Modulation by early-life infection. J Neurosci 31: 15511–15521. doi: 10.1523/JNEUROSCI.3688-11.2011
|
| [49] |
Caraci F, Gulisano W, Guida CA, et al. (2015) A key role for TGF-β1 in hippocampal synaptic plasticity and memory. Sci Rep 5: 11252. doi: 10.1038/srep11252
|
| [50] |
Butovsky O, Jedrychowski MP, Moore CS, et al. (2014) Identification of a unique TGF-β-dependent molecular and functional signature in microglia. Nat Neurosci 17: 131–143. doi: 10.1038/nn.3599
|
| [51] |
Sanes JR, Yamagata M (2009) Many paths to synaptic specificity. Annu Rev Cell Dev Biol 25: 161–195. doi: 10.1146/annurev.cellbio.24.110707.175402
|
| [52] |
Holtmaat A, Svoboda K (2009) Experience-dependent structural synaptic plasticity in the mammalian brain. Nat Rev Neurosci 10: 647–658. doi: 10.1038/nrn2699
|
| [53] |
Holtmaat AJGD, Trachtenberg JT, Wilbrecht L, et al. (2005) Transient and persistent dendritic spines in the neocortex in vivo. Neuron 45: 279–291. doi: 10.1016/j.neuron.2005.01.003
|
| [54] |
Caroni P, Donato F, Muller D (2012) Structural plasticity upon learning: Regulation and functions. Nat Rev Neurosci 13: 478–490. doi: 10.1038/nrn3258
|
| [55] |
Blinzinger K, Kreutzberg G (1968) Displacement of synaptic terminals from regenerating motoneurons by microglial cells. Z Zellforsch Mikrosk Anat 85: 145–157. doi: 10.1007/BF00325030
|
| [56] |
Trapp BD, Wujek JR, Criste GA, et al. (2007) Evidence for synaptic stripping by cortical microglia. Glia 55: 360–368. doi: 10.1002/glia.20462
|
| [57] |
Wake H, Moorhouse AJ, Jinno S, et al. (2009) Resting microglia directly monitor the functional state of synapses in vivo and determine the fate of ischemic terminals. J Neurosci 29: 3974–3980. doi: 10.1523/JNEUROSCI.4363-08.2009
|
| [58] |
Tremblay Mè, Lowery RL, Majewska AK (2010) Microglial interactions with synapses are modulated by visual experience. PLoS Biol 8: e1000527. doi: 10.1371/journal.pbio.1000527
|
| [59] |
Ji K, Akgul G, Wollmuth LP, et al. (2013) Microglia actively regulate the number of functional synapses. PLoS One 8: e56293. doi: 10.1371/journal.pone.0056293
|
| [60] |
Torres L, Danver J, Ji K, et al. (2016) Dynamic microglial modulation of spatial learning and social behavior. Brain Behav Immun 55: 6–16. doi: 10.1016/j.bbi.2015.09.001
|
| [61] |
Lowery RL, Tremblay Mè, Hopkins BE, et al. (2017) The microglial fractalkine receptor is not required for activity-dependent plasticity in the mouse visual system. Glia 65: 1744–1761. doi: 10.1002/glia.23192
|
| [62] |
Dalmau I, Finsen B, Zimmer J, et al. (1998) Development of microglia in the postnatal rat hippocampus. Hippocampus 8: 458–474. doi: 10.1002/(SICI)1098-1063(1998)8:5<458::AID-HIPO6>3.0.CO;2-N
|
| [63] |
Squarzoni P, Oller G, Hoeffel G, et al. (2014) Microglia modulate wiring of the embryonic forebrain. Cell Rep 8: 1271–1279. doi: 10.1016/j.celrep.2014.07.042
|
| [64] |
Bessis A, Béchade C, Bernard D, et al. (2007) Microglial control of neuronal death and synaptic properties. Glia 55: 233–238. doi: 10.1002/glia.20459
|
| [65] |
Parkhurst CN, Yang G, Ninan I, et al. (2013) Microglia promote learning-dependent synapse formation through brain-derived neurotrophic factor. Cell 155: 1596–1609. doi: 10.1016/j.cell.2013.11.030
|
| [66] |
Bennett ML, Bennett FC, Liddelow SA, et al. (2016) New tools for studying microglia in the mouse and human CNS. Proc Natl Acad Sci U S A 113: E1738–E1746. doi: 10.1073/pnas.1525528113
|
| [67] |
Miyamoto A, Wake H, Ishikawa AW, et al. (2016) Microglia contact induces synapse formation in developing somatosensory cortex. Nat Commun 7: 12540. doi: 10.1038/ncomms12540
|
| [68] |
Chugh D, Nilsson P, Afjei SA, et al. (2013) Brain inflammation induces post-synaptic changes during early synapse formation in adult-born hippocampal neurons. Exp Neurol 250: 176–188. doi: 10.1016/j.expneurol.2013.09.005
|
| [69] | Cardona SM, Mendiola AS, Yang YC, et al. (2015) Disruption of fractalkine signaling leads to microglial activation and neuronal damage in the diabetic retina. ASN Neuro 7: 1–18. |
| [70] |
Moran LB, Graeber MB (2004) The facial nerve axotomy model. Brain Res Rev 44: 154–178. doi: 10.1016/j.brainresrev.2003.11.004
|
| [71] |
Chamak B, Dobbertin A, Mallat M (1995) Immunohistochemical detection of thrombospondin in microglia in the developing rat brain. Neuroscience 69: 177–187. doi: 10.1016/0306-4522(95)00236-C
|
| [72] |
Christopherson KS, Ullian EM, Stokes CCA, et al. (2005) Thrombospondins are astrocyte-secreted proteins that promote CNS synaptogenesis. Cell 120: 421–433. doi: 10.1016/j.cell.2004.12.020
|
| [73] |
M?ller JC, Klein MA, Haas S, et al. (1996) Regulation of thrombospondin in the regenerating mouse facial motor nucleus. Glia 17: 121–132. doi: 10.1002/(SICI)1098-1136(199606)17:2<121::AID-GLIA4>3.0.CO;2-5
|
| [74] |
Morris GP, Clark IA, Zinn R, et al. (2013) Microglia: A new frontier for synaptic plasticity, learning and memory, and neurodegenerative disease research. Neurobiol Learn Mem 105: 40–53. doi: 10.1016/j.nlm.2013.07.002
|
| [75] | Schwarz JM, Sholar PW, Bilbo SD (2012) Sex differences in microglial colonization of the developing rat brain. J Neurochem 120: 948–963. |
| [76] |
Lenz KM, Nugent BM, Haliyur R, et al. (2013) Microglia are essential to masculinization of brain and behavior. J Neurosci 33: 2761–2772. doi: 10.1523/JNEUROSCI.1268-12.2013
|
| [77] |
Sorge RE, Mapplebeck JCS, Rosen S, et al. (2015) Different immune cells mediate mechanical pain hypersensitivity in male and female mice. Nat Neurosci 18: 1081–1083. doi: 10.1038/nn.4053
|
| [78] |
Wu MV, Shah NM (2011) Control of masculinization of the brain and behavior. Curr Opin Neurobiol 21: 116–123. doi: 10.1016/j.conb.2010.09.014
|
| [79] |
Mendez P, Garcia-Segura LM, Muller D (2011) Estradiol promotes spine growth and synapse formation without affecting pre-established networks. Hippocampus 21: 1263–1267. doi: 10.1002/hipo.20875
|
| [80] |
Nelson LH, Warden S, Lenz KM (2017) Sex differences in microglial phagocytosis in the neonatal hippocampus. Brain Behav Immun 64: 11–22. doi: 10.1016/j.bbi.2017.03.010
|
| [81] |
Bettis TJ, Jacobs LF (2009) Sex-specific strategies in spatial orientation in C57BL/6J mice. Behav Processes 82: 249–255. doi: 10.1016/j.beproc.2009.07.004
|
| [82] |
Cahill L (2006) Why sex matters for neuroscience. Nat Rev Neurosci 7: 477–484. doi: 10.1038/nrn1909
|
| [83] |
Rademakers R, Baker M, Nicholson AM, et al. (2012) Mutations in the colony stimulating factor 1 receptor (CSF1R) gene cause hereditary diffuse leukoencephalopathy with spheroids. Nat Genet 44: 200–205. doi: 10.1038/ng.1027
|
| [84] |
Bianchin MM, Capella HM, Chaves DL, et al. (2004) Nasu-Hakola disease (polycystic lipomembranous osteodysplasia with sclerosing leukoencephalopathy-PLOSL): A dementia associated with bone cystic lesions. Cell Mol Neurobiol 24: 1–24. doi: 10.1023/B:CEMN.0000012721.08168.ee
|
| [85] |
Paloneva J, Kestil? M, Wu J, et al. (2000) Loss-of-function mutations in TYROBP (DAP12) result in a presenile dementia with bone cysts. Nat Genet 25: 357–361. doi: 10.1038/77153
|
| [86] |
Paloneva J, Manninen T, Christman G, et al. (2002) Mutations in two genes encoding different subunits of a receptor signaling complex result in an identical disease phenotype. Am J Hum Genet 71: 656–662. doi: 10.1086/342259
|
| [87] | Hakola HP (1972) Neuropsychiatric and genetic aspects of a new hereditary disease characterized by progressive dementia and lipomembranous polycystic osteodysplasia. Acta Psychiatr Scand Suppl 232: 1–173. |
| [88] | Nasu T, Tsukahara Y, Terayama K (1973) A lipid metabolic disease-"membranous lipodystrophy" -an autopsy case demonstrating numerous peculiar membrane-structures composed of compound lipid in bone and bone marrow and various adipose tissues. Acta Pathol Jpn 23: 539–558. |
| [89] |
Takahashi K, Rochford CDP, Neumann H (2005) Clearance of apoptotic neurons without inflammation by microglial triggering receptor expressed on myeloid cells-2. J Exp Med 201: 647–657. doi: 10.1084/jem.20041611
|
| [90] |
Wakselman S, Béchade C, Roumier A, et al. (2008) Developmental neuronal death in hippocampus requires the microglial CD11b integrin and DAP12 immunoreceptor. J Neurosci 28: 8138–8143. doi: 10.1523/JNEUROSCI.1006-08.2008
|
| [91] | Axelsson R, R?ytt? M, Sourander P, et al. (1984) Hereditary diffuse leucoencephalopathy with spheroids. Acta Psychiatr Scand Suppl 314: 1–65. |
| [92] |
Elmore MRP, Najafi AR, Koike MA, et al. (2014) Colony-stimulating factor 1 receptor signaling is necessary for microglia viability, unmasking a microglia progenitor cell in the adult brain. Neuron 82: 380–397. doi: 10.1016/j.neuron.2014.02.040
|
| [93] | Erblich B, Zhu L, Etgen AM, et al. (2011) Absence of colony stimulation factor-1 receptor results in loss of microglia, disrupted brain development and olfactory deficits. PLoS One 6: e26317. |
| [94] | Greter M, Lelios I, Pelczar P, et al. (2012) Stroma-derived interleukin-34 controls the development and maintenance of langerhans cells and the maintenance of microglia. Immunity 37: 1050–1060. |
| [95] | Wang Y, Szretter KJ, Vermi W, et al. (2012) IL-34 is a tissue-restricted ligand of CSF1R required for the development of Langerhans cells and microglia. Nat Immunol 13: 753–760. |
| [96] | Otero K, Turnbull IR, Poliani PL, et al. (2009) Macrophage colony-stimulating factor induces the proliferation and survival of macrophages via a pathway involving DAP12 and beta-catenin. Nat Immunol 10: 734–743. |
| [97] |
Konno T, Tada M, Tada M, et al. (2014) Haploinsufficiency of CSF-1R and clinicopathologic characterization in patients with HDLS. Neurology 82: 139–148. doi: 10.1212/WNL.0000000000000046
|
| [98] |
Sundal C, Lash J, Aasly J, et al. (2012) Hereditary diffuse leukoencephalopathy with axonal spheroids (HDLS): A misdiagnosed disease entity. J Neurol Sci 314: 130–137. doi: 10.1016/j.jns.2011.10.006
|
| [99] |
Matcovitch-Natan O, Winter DR, Giladi A, et al. (2016) Microglia development follows a stepwise program to regulate brain homeostasis. Science 353: aad8670. doi: 10.1126/science.aad8670
|
| [100] |
Schaafsma W, Basterra LB, Jacobs S, et al. (2017) Maternal inflammation induces immune activation of fetal microglia and leads to disrupted microglia immune responses, behavior, and learning performance in adulthood. Neurobiol Dis 106: 291–300. doi: 10.1016/j.nbd.2017.07.017
|
| [101] |
Bilbo SD, Levkoff LH, Mahoney JH, et al. (2005) Neonatal infection induces memory impairments following an immune challenge in adulthood. Behav Neurosci 119: 293–301. doi: 10.1037/0735-7044.119.1.293
|
| [102] |
Bilbo SD, Biedenkapp JC, Der-Avakian A, et al. (2005) Neonatal infection-induced memory impairment after lipopolysaccharide in adulthood is prevented via caspase-1 inhibition. J Neurosci 25: 8000–8009. doi: 10.1523/JNEUROSCI.1748-05.2005
|
| [103] |
Osborne BF, Caulfield JI, Solomotis SA, et al. (2017) Neonatal infection produces significant changes in immune function with no associated learning deficits in juvenile rats. Dev Neurobiol 77: 1221–1236. doi: 10.1002/dneu.22512
|
| [104] |
Knuesel I, Chicha L, Britschgi M, et al. (2014) Maternal immune activation and abnormal brain development across CNS disorders. Nat Rev Neurol 10: 643–660. doi: 10.1038/nrneurol.2014.187
|
| [105] |
Boitard C, Cavaroc A, Sauvant J, et al. (2014) Impairment of hippocampal-dependent memory induced by juvenile high-fat diet intake is associated with enhanced hippocampal inflammation in rats. Brain Behav Immun 40: 9–17. doi: 10.1016/j.bbi.2014.03.005
|
| [106] |
Hsu TM, Konanur VR, Taing L, et al. (2015) Effects of sucrose and high fructose corn syrup consumption on spatial memory function and hippocampal neuroinflammation in adolescent rats. Hippocampus 25: 227–239. doi: 10.1002/hipo.22368
|
| [107] |
De Luca SN, Ziko I, Sominsky L, et al. (2016) Early life overfeeding impairs spatial memory performance by reducing microglial sensitivity to learning. J Neuroinflammation 13: 1–15. doi: 10.1186/s12974-015-0467-5
|
| [108] |
Hao S, Dey A, Yu X, et al. (2016) Dietary obesity reversibly induces synaptic stripping by microglia and impairs hippocampal plasticity. Brain Behav Immun 51: 230–239. doi: 10.1016/j.bbi.2015.08.023
|
| [109] |
Tanaka S, Ide M, Shibutani T, et al. (2006) Lipopolysaccharide-induced microglial activation induces learning and memory deficits without neuronal cell death in rats. J Neurosci Res 83: 557–566. doi: 10.1002/jnr.20752
|
| [110] |
Riazi K, Galic MA, Kuzmiski JB, et al. (2008) Microglial activation and TNFalpha production mediate altered CNS excitability following peripheral inflammation. Proc Natl Acad Sci U.S.A 105: 17151–17156. doi: 10.1073/pnas.0806682105
|
| [111] |
Riazi K, Galic MA, Kentner AC, et al. (2015) Microglia-dependent alteration of glutamatergic synaptic transmission and plasticity in the hippocampus during peripheral inflammation. J Neurosci 35: 4942–4952. doi: 10.1523/JNEUROSCI.4485-14.2015
|
| [112] |
D'Mello C, Riazi K, Le T, et al. (2013) P-selectin-mediated monocyte-cerebral endothelium adhesive interactions link peripheral organ inflammation to sickness behaviors. J Neurosci 33: 14878–14888. doi: 10.1523/JNEUROSCI.1329-13.2013
|
| [113] |
Vasek MJ, Garber C, Dorsey D, et al. (2016) A complement-microglial axis drives synapse loss during virus-induced memory impairment. Nature 534: 538–543. doi: 10.1038/nature18283
|
| [114] |
Minami M, Kuraishi Y, Yamaguchi T, et al. (1991) Immobilization stress induces interleukin-1 beta mRNA in the rat hypothalamus. Neurosci Lett 123: 254–256. doi: 10.1016/0304-3940(91)90944-O
|
| [115] | Nguyen KT, Deak T, Owens SM, et al. (1998) Exposure to acute stress induces brain interleukin-1beta protein in the rat. J Neurosci 18: 2239–2246. |
| [116] |
Pugh CR, Nguyen KT, Gonyea JL, et al. (1999) Role of interleukin-1 beta in impairment of contextual fear conditioning caused by social isolation. Behav Brain Res 106: 109–118. doi: 10.1016/S0166-4328(99)00098-4
|
| [117] |
Sugama S, Fujita M, Hashimoto M, et al. (2007) Stress induced morphological microglial activation in the rodent brain: Involvement of interleukin-18. Neuroscience 146: 1388–1399. doi: 10.1016/j.neuroscience.2007.02.043
|
| [118] |
Li S, Wang C, Wang W, et al. (2008) Chronic mild stress impairs cognition in mice: From brain homeostasis to behavior. Life Sci 82: 934–942. doi: 10.1016/j.lfs.2008.02.010
|
| [119] |
Frank MG, Baratta MV, Sprunger DB, et al. (2007) Microglia serve as a neuroimmune substrate for stress-induced potentiation of CNS pro-inflammatory cytokine responses. Brain Behav Immun 21: 47–59. doi: 10.1016/j.bbi.2006.03.005
|
| [120] |
Ohgidani M, Kato TA, Sagata N, et al. (2016) TNF-α from hippocampal microglia induces working memory deficits by acute stress in mice. Brain Behav Immun 55: 17–24. doi: 10.1016/j.bbi.2015.08.022
|
| [121] |
Hinwood M, Morandini J, Day TA, et al. (2012) Evidence that microglia mediate the neurobiological effects of chronic psychological stress on the medial prefrontal cortex. Cereb Cortex 22: 1442–1454. doi: 10.1093/cercor/bhr229
|
| [122] |
Kreisel T, Frank MG, Licht T, et al. (2014) Dynamic microglial alterations underlie stress-induced depressive-like behavior and suppressed neurogenesis. Mol Psychiatry 19: 699–709. doi: 10.1038/mp.2013.155
|
| [123] |
Sugama S (2009) Stress-induced microglial activation may facilitate the progression of neurodegenerative disorders. Med Hypotheses 73: 1031–1034. doi: 10.1016/j.mehy.2009.02.047
|
| [124] | Diz-Chaves Y, Pernía O, Carrero P, et al. (2012) Prenatal stress causes alterations in the morphology of microglia and the inflammatory response of the hippocampus of adult female mice. J Neuroinflammation 9: 1–10. |
| [125] |
Frank MG, Thompson BM, Watkins LR, et al. (2012) Glucocorticoids mediate stress-induced priming of microglial pro-inflammatory responses. Brain Behav Immun 26: 337–345. doi: 10.1016/j.bbi.2011.10.005
|
| [126] |
Jauregui-Huerta F, Ruvalcaba-Delgadillo Y, Gonzalez-Casta?eda R, et al. (2010) Responses of glial cells to stress and glucocorticoids. Curr Immunol Rev 6: 195–204. doi: 10.2174/157339510791823790
|
| [127] |
Dantzer R, O'Connor JC, Freund GG, et al. (2008) From inflammation to sickness and depression: When the immune system subjugates the brain. Nat Rev Neurosci 9: 46–56. doi: 10.1038/nrn2297
|
| [128] |
Bachtell R, Hutchinson MR, Wang X, et al. (2015) Targeting the toll of drug abuse: The translational potential of Toll-like receptor 4. CNS Neurol Disord Drug Targets 14: 692–699. doi: 10.2174/1871527314666150529132503
|
| [129] | Nadjar A, Wigren HKM, Tremblay Mè (2017) Roles of microglial phagocytosis and inflammatory mediators in the pathophysiology of sleep disorders. Front Cell Neurosci 11: 250. |
| [130] |
Liu MC, Liu XQ, Wang W, et al. (2012) Involvement of microglia activation in the lead induced long-term potentiation impairment. PLoS One 7: e43924. doi: 10.1371/journal.pone.0043924
|
| [131] |
Akinrinade ID, Memudu AE, Ogundele OM, et al. (2015) Interplay of glia activation and oxidative stress formation in fluoride and aluminium exposure. Pathophysiology 22: 39–48. doi: 10.1016/j.pathophys.2014.12.001
|
| [132] |
Hefendehl JK, Neher JJ, Sühs RB, et al. (2014) Homeostatic and injury-induced microglia behavior in the aging brain. Aging Cell 13: 60–69. doi: 10.1111/acel.12149
|
| [133] |
Ritzel RM, Patel AR, Pan S, et al. (2015) Age- and location-related changes in microglial function. Neurobiol Aging 36: 2153–2163. doi: 10.1016/j.neurobiolaging.2015.02.016
|
| [134] |
Streit WJ, Sammons NW, Kuhns AJ, et al. (2004) Dystrophic microglia in the aging human brain. Glia 45: 208–212. doi: 10.1002/glia.10319
|
| [135] |
Streit WJ, Braak H, Xue QS, et al. (2009) Dystrophic (senescent) rather than activated microglial cells are associated with tau pathology and likely precede neurodegeneration in Alzheimer's disease. Acta Neuropathol 118: 475–485. doi: 10.1007/s00401-009-0556-6
|
| [136] |
Tremblay Mè, Zettel ML, Ison JR, et al. (2012) Effects of aging and sensory loss on glial cells in mouse visual and auditory cortices. Glia 60: 541–558. doi: 10.1002/glia.22287
|
| [137] | Njie EG, Boelen E, Stassen FR, et al. (2012) Ex vivo cultures of microglia from young and aged rodent brain reveal age-related changes in microglial function. Neurobiol Aging 33: 195.e1–195.e12. |
| [138] |
Bisht K, Sharma KP, Lecours C, et al. (2016) Dark microglia: A new phenotype predominantly associated with pathological states. Glia 64: 826–839. doi: 10.1002/glia.22966
|
| [139] | Mosher KI, Wyss-Coray T (2014) Microglial dysfunction in brain aging and Alzheimer's disease. Biochem Pharmacol 88: 594–604. |
| [140] |
Sierra A, Gottfried-Blackmore AC, McEwen BS, et al. (2007) Microglia derived from aging mice exhibit an altered inflammatory profile. Glia 55: 412–424. doi: 10.1002/glia.20468
|
| [141] |
Stichel CC, Luebbert H (2007) Inflammatory processes in the aging mouse brain: Participation of dendritic cells and T-cells. Neurobiol Aging 28: 1507–1521. doi: 10.1016/j.neurobiolaging.2006.07.022
|
| [142] |
Füger P, Hefendehl JK, Veeraraghavalu K, et al. (2017) Microglia turnover with aging and in an Alzheimer's model via long-term in vivo single-cell imaging. Nat Neurosci 20: 1371–1376. doi: 10.1038/nn.4631
|
| [143] |
Cartier N, Lewis CA, Zhang R, et al. (2014) The role of microglia in human disease: Therapeutic tool or target? Acta Neuropathol 128: 363–380. doi: 10.1007/s00401-014-1330-y
|
| [144] |
Cunningham C (2013) Microglia and neurodegeneration: The role of systemic inflammation. Glia 61: 71–90. doi: 10.1002/glia.22350
|
| [145] |
Perry VH, Holmes C (2014) Microglial priming in neurodegenerative disease. Nat Rev Neurol 10: 217–224. doi: 10.1038/nrneurol.2014.38
|
| [146] |
Solano Fonseca R, Mahesula S, Apple DM, et al. (2016) Neurogenic niche microglia undergo positional remodeling and progressive activation contributing to age-associated reductions in neurogenesis. Stem Cells Dev 25: 542–555. doi: 10.1089/scd.2015.0319
|
| [147] |
Shi Q, Colodner KJ, Matousek SB, et al. (2015) Complement C3-deficient mice fail to display age-related hippocampal decline. J Neurosci 35: 13029–13042. doi: 10.1523/JNEUROSCI.1698-15.2015
|
| [148] |
Querfurth HW, LaFerla FM (2010) Alzheimer's disease. N Engl J Med 362: 329–344. doi: 10.1056/NEJMra0909142
|
| [149] |
Greenberg SG, Davies P (1990) A preparation of Alzheimer paired helical filaments that displays distinct tau proteins by polyacrylamide gel electrophoresis. Proc Natl Acad Sci U.S.A 87: 5827–5831. doi: 10.1073/pnas.87.15.5827
|
| [150] | Serrano-Pozo A, Frosch MP, Masliah E, et al. (2011) Neuropathological alterations in Alzheimer disease. Cold Spring Harb Perspect Med 1: a006189. |
| [151] |
Akiyama H, Barger S, Barnum S, et al. (2000) Inflammation and Alzheimer's disease. Neurobiol Aging 21: 383–421. doi: 10.1016/S0197-4580(00)00124-X
|
| [152] |
Hardy J, Selkoe DJ (2002) The amyloid hypothesis of Alzheimer's disease: Progress and problems on the road to therapeutics. Science 297: 353–356. doi: 10.1126/science.1072994
|
| [153] |
Alzheimer A, Stelzmann RA, Schnitzlein HN, et al. (1995) An English translation of Alzheimer's 1907 paper, "Uber eine eigenartige Erkankung der Hirnrinde". Clin Anat 8: 429–431. doi: 10.1002/ca.980080612
|
| [154] |
Bolmont T, Haiss F, Eicke D, et al. (2008) Dynamics of the microglial/amyloid interaction indicate a role in plaque maintenance. J Neurosci 28: 4283–4292. doi: 10.1523/JNEUROSCI.4814-07.2008
|
| [155] |
Meyer-Luehmann M, Spires-Jones TL, Prada C, et al. (2008) Rapid appearance and local toxicity of amyloid-β plaques in a mouse model of Alzheimer's disease. Nature 451: 720–724. doi: 10.1038/nature06616
|
| [156] |
Wisniewski HM, Wegiel J, Wang KC, et al. (1989) Ultrastructural studies of the cells forming amyloid fibers in classical plaques. Can J Neurol Sci 16: 535–542. doi: 10.1017/S0317167100029887
|
| [157] |
Bornemann KD, Wiederhold KH, Pauli C, et al. (2001) Abeta-induced inflammatory processes in microglia cells of APP23 transgenic mice. Amer J Pathol 158: 63–73. doi: 10.1016/S0002-9440(10)63945-4
|
| [158] | Floden AM, Combs CK (2011) Microglia demonstrate age-dependent interaction with amyloid-β fibrils. J Alzheimers Dis 25: 279–293. |
| [159] |
Hickman SE, Allison EK, Khoury JE (2008) Microglial dysfunction and defective beta-amyloid clearance pathways in aging Alzheimer's disease mice. J Neurosci 28: 8354–8360. doi: 10.1523/JNEUROSCI.0616-08.2008
|
| [160] |
Paolicelli RC, Jawaid A, Henstridge CM, et al. (2017) TDP-43 depletion in microglia promotes amyloid clearance but also induces synapse loss. Neuron 95: 297–308. doi: 10.1016/j.neuron.2017.05.037
|
| [161] |
Hong S, Beja-Glasser VF, Nfonoyim BM, et al. (2016) Complement and microglia mediate early synapse loss in Alzheimer mouse models. Science 352: 712–716. doi: 10.1126/science.aad8373
|
| [162] |
Shi Q, Chowdhury S, Ma R, et al. (2017) Complement C3 deficiency protects against neurodegeneration in aged plaque-rich APP/PS1 mice. Sci Transl Med 9: eaaf6295. doi: 10.1126/scitranslmed.aaf6295
|
| [163] |
Bertram L, Lange C, Mullin K, et al. (2008) Genome-wide association analysis reveals putative Alzheimer's disease susceptibility loci in addition to APOE. Am J Hum Genet 83: 623–632. doi: 10.1016/j.ajhg.2008.10.008
|
| [164] |
Lambert JC, Heath S, Even G, et al. (2009) Genome-wide association study identifies variants at CLU and CR1 associated with Alzheimer's disease. Nat Genet 41: 1094–1099. doi: 10.1038/ng.439
|
| [165] |
Lambert JC, Ibrahim-Verbaas CA, Harold D, et al. (2013) Meta-analysis of 74,046 individuals identifies 11 new susceptibility loci for Alzheimer's disease. Nat Genet 45: 1452–1458. doi: 10.1038/ng.2802
|
| [166] |
Jonsson T, Stefansson H, Steinberg S, et al. (2013) Variant of TREM2 associated with the risk of Alzheimer's disease. N Engl J Med 368: 107–116. doi: 10.1056/NEJMoa1211103
|
| [167] |
Naj AC, Jun G, Beecham GW, et al. (2011) Common variants at MS4A4/MS4A6E, CD2AP, CD33 and EPHA1 are associated with late-onset Alzheimer's disease. Nat Genet 43: 436–441. doi: 10.1038/ng.801
|
| [168] |
Zhang B, Gaiteri C, Bodea LG, et al. (2013) Integrated systems approach identifies genetic nodes and networks in late-onset Alzheimer's disease. Cell 153: 707–720. doi: 10.1016/j.cell.2013.03.030
|
| [169] |
Frank S, Burbach GJ, Bonin M, et al. (2008) TREM2 is upregulated in amyloid plaque-associated microglia in aged APP23 transgenic mice. Glia 56: 1438–1447. doi: 10.1002/glia.20710
|
| [170] |
Guerreiro R, Wojtas A, Bras J, et al. (2013) TREM2 variants in Alzheimer's disease. N Engl J Med 368: 117–127. doi: 10.1056/NEJMoa1211851
|
| [171] |
Keren-Shaul H, Spinrad A, Weiner A, et al. (2017) A unique microglia type associated with restricting development of Alzheimer's disease. Cell 169: 1276. doi: 10.1016/j.cell.2017.05.018
|
| [172] | Melchior B, Garcia AE, Hsiung BK, et al. (2010) Dual induction of TREM2 and tolerance-related transcript, Tmem176b, in amyloid transgenic mice: Implications for vaccine-based therapies for Alzheimer's disease. ASN Neuro 2: e00037. |
| [173] |
Krasemann S, Madore C, Cialic R, et al. (2017) The TREM2-APOE pathway drives the transcriptional phenotype of dysfunctional microglia in neurodegenerative diseases. Immunity 47: 566–581. doi: 10.1016/j.immuni.2017.08.008
|
| [174] |
Wang Y, Ulland TK, Ulrich JD, et al. (2016) TREM2-mediated early microglial response limits diffusion and toxicity of amyloid plaques. J Exp Med 213: 667–675. doi: 10.1084/jem.20151948
|
| [175] |
Yuan P, Condello C, Keene CD, et al. (2016) TREM2 haplodeficiency in mice and humans impairs the microglia barrier function leading to decreased amyloid compaction and severe axonal dystrophy. Neuron 90: 724–739. doi: 10.1016/j.neuron.2016.05.003
|
| [176] |
Kleinberger G, Brendel M, Mracsko E, et al. (2017) The FTD-like syndrome causing TREM2 T66M mutation impairs microglia function, brain perfusion, and glucose metabolism. EMBO J 36: 1837–1853. doi: 10.15252/embj.201796516
|
| [177] |
Jay TR, Hirsch AM, Broihier ML, et al. (2017) Disease progression-dependent effects of TREM2 deficiency in a mouse model of Alzheimer's disease. J Neurosci 37: 637–647. doi: 10.1523/JNEUROSCI.2110-16.2016
|
| [178] |
Wang Y, Cella M, Mallinson K, et al. (2015) TREM2 lipid sensing sustains the microglial response in an Alzheimer's disease model. Cell 160: 1061–1071. doi: 10.1016/j.cell.2015.01.049
|
| [179] |
Jay TR, Miller CM, Cheng PJ, et al. (2015) TREM2 deficiency eliminates TREM2+ inflammatory macrophages and ameliorates pathology in Alzheimer's disease mouse models. J Exp Med 212: 287–295. doi: 10.1084/jem.20142322
|
| [180] |
Ulrich JD, Finn MB, Wang Y, et al. (2014) Altered microglial response to Aβ plaques in APPPS1-21 mice heterozygous for TREM2. Mol Neurodegener 9: 1–9. doi: 10.1186/1750-1326-9-1
|
| [181] |
Ulland TK, Song WM, Huang SCC, et al. (2017) TREM2 maintains microglial metabolic fitness in Alzheimer's disease. Cell 170: 649. doi: 10.1016/j.cell.2017.07.023
|
| [182] | Mazaheri F, Snaidero N, Kleinberger G, et al. (2017) TREM2 deficiency impairs chemotaxis and microglial responses to neuronal injury. EMBO Rep 18: 1186–1198. |
| [183] |
Leyns CEG, Ulrich JD, Finn MB, et al. (2017) TREM2 deficiency attenuates neuroinflammation and protects against neurodegeneration in a mouse model of tauopathy. Proc Natl Acad Sci U.S.A 114: 11524–11529. doi: 10.1073/pnas.1710311114
|
| [184] |
Fuhrmann M, Bittner T, Jung CKE, et al. (2010) Microglial Cx3cr1 knockout prevents neuron loss in a mouse model of Alzheimer's disease. Nat Neurosci 13: 411–413. doi: 10.1038/nn.2511
|
| [185] |
Lee S, Varvel NH, Konerth ME, et al. (2010) CX3CR1 deficiency alters microglial activation and reduces beta-amyloid deposition in two Alzheimer's disease mouse models. Amer J Pathol 177: 2549–2562. doi: 10.2353/ajpath.2010.100265
|
| [186] |
Liu Z, Condello C, Schain A, et al. (2010) CX3CR1 in microglia regulates brain amyloid deposition through selective protofibrillar amyloid-β phagocytosis. J Neurosci 30: 17091–17101. doi: 10.1523/JNEUROSCI.4403-10.2010
|
| [187] |
Bhaskar K, Konerth M, Kokiko-Cochran ON, et al. (2010) Regulation of tau pathology by the microglial fractalkine receptor. Neuron 68: 19–31. doi: 10.1016/j.neuron.2010.08.023
|
| [188] |
Cho SH, Sun B, Zhou Y, et al. (2011) CX3CR1 protein signaling modulates microglial activation and protects against plaque-independent cognitive deficits in a mouse model of Alzheimer disease. J Biol Chem 286: 32713–32722. doi: 10.1074/jbc.M111.254268
|
| [189] |
Maphis N, Xu G, Kokiko-Cochran ON, et al. (2015) Reactive microglia drive tau pathology and contribute to the spreading of pathological tau in the brain. Brain 138: 1738–1755. doi: 10.1093/brain/awv081
|
| [190] |
Diaz-Aparicio I, Beccari S, Abiega O, et al. (2016) Clearing the corpses: Regulatory mechanisms, novel tools, and therapeutic potential of harnessing microglial phagocytosis in the diseased brain. Neural Regen Res 11: 1533–1539. doi: 10.4103/1673-5374.193220
|
| [191] |
Abiega O, Beccari S, Diaz-Aparicio I, et al. (2016) Neuronal hyperactivity disturbs ATP microgradients, impairs microglial motility, and reduces phagocytic receptor expression triggering apoptosis/microglial phagocytosis uncoupling. PLoS Biol 14: e1002466. doi: 10.1371/journal.pbio.1002466
|
| [192] |
Qian Z, Gilbert ME, Colicos MA, et al. (1993) Tissue-plasminogen activator is induced as an immediate-early gene during seizure, kindling and long-term potentiation. Nature 361: 453–457. doi: 10.1038/361453a0
|
| [193] |
Wu YP, Siao CJ, Lu W, et al. (2000) The tissue plasminogen activator (tPA)/plasmin extracellular proteolytic system regulates seizure-induced hippocampal mossy fiber outgrowth through a proteoglycan substrate. J Cell Biol 148: 1295–1304. doi: 10.1083/jcb.148.6.1295
|
| [194] |
Abraham J, Fox PD, Condello C, et al. (2012) Minocycline attenuates microglia activation and blocks the long-term epileptogenic effects of early-life seizures. Neurobiol Dis 46: 425–430. doi: 10.1016/j.nbd.2012.02.006
|
| [195] |
Wang N, Mi X, Gao B, et al. (2015) Minocycline inhibits brain inflammation and attenuates spontaneous recurrent seizures following pilocarpine-induced status epilepticus. Neuroscience 287: 144–156. doi: 10.1016/j.neuroscience.2014.12.021
|
| [196] |
Nowak M, Strzelczyk A, Reif PS, et al. (2012) Minocycline as potent anticonvulsant in a patient with astrocytoma and drug resistant epilepsy. Seizure 21: 227–228. doi: 10.1016/j.seizure.2011.12.009
|
| [197] |
Gass J, Prudencio M, Stetler C, et al. (2012) Progranulin: An emerging target for FTLD therapies. Brain Res 1462: 118–128. doi: 10.1016/j.brainres.2012.01.047
|
| [198] |
Krabbe G, Minami SS, Etchegaray JI, et al. (2017) Microglial NFκB-TNFα hyperactivation induces obsessive-compulsive behavior in mouse models of progranulin-deficient frontotemporal dementia. Proc Natl Acad Sci U S A 114: 5029–5034. doi: 10.1073/pnas.1700477114
|
| [199] | DeJesus-Hernandez M, Mackenzie IR, Boeve BF, et al. (2011) Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron 72: 245–256. |
| [200] | Vance C, Al-Chalabi A, Ruddy D, et al. (2006) Familial amyotrophic lateral sclerosis with frontotemporal dementia is linked to a locus on chromosome 9p13.2–21.3. Brain 129: 868–876. |
| [201] | Borroni B, Ferrari F, Galimberti D, et al. (2014) Heterozygous TREM2 mutations in frontotemporal dementia. Neurobiol Aging 35: 7–10. |
| [202] | Guerreiro R, Bilgic B, Guven G, et al. (2013) Novel compound heterozygous mutation in TREM2 found in a Turkish frontotemporal dementia-like family. Neurobiol Aging 34: 2890.e1–2890.e5. |
| [203] | Le Ber I, De Septenville A, Guerreiro R, et al. (2014) Homozygous TREM2 mutation in a family with atypical frontotemporal dementia. Neurobiol Aging 35: 2419.e23–2419.e25. |
| [204] | Thelen M, Razquin C, Hernández I, et al. (2014) Investigation of the role of rare TREM2 variants in frontotemporal dementia subtypes. Neurobiol Aging 35: 2657.e13–2657.e19. |
| [205] |
Mackenzie IR, Rademakers R (2008) The role of transactive response DNA-binding protein-43 in amyotrophic lateral sclerosis and frontotemporal dementia. Curr Opin Neurol 21: 693–700. doi: 10.1097/WCO.0b013e3283168d1d
|
| [206] |
Neumann M, Sampathu DM, Kwong LK, et al. (2006) Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science 314: 130–133. doi: 10.1126/science.1134108
|
| [207] |
Xia Q, Hu Q, Wang H, et al. (2015) Induction of COX-2-PGE2 synthesis by activation of the MAPK/ERK pathway contributes to neuronal death triggered by TDP-43-depleted microglia. Cell Death Dis 6: e1702. doi: 10.1038/cddis.2015.69
|
| [208] | Benitez BA, Cooper B, Pastor P, et al. (2013) TREM2 is associated with the risk of Alzheimer's disease in Spanish population. Neurobiol Aging 34: 1711.e15–1711.e17. |
| [209] | Rayaprolu S, Mullen B, Baker M, et al. (2013) TREM2 in neurodegeneration: Evidence for association of the p.R47H variant with frontotemporal dementia and Parkinson's disease. Mol Neurodegener 8: 19. |
| [210] |
Gerhard A, Pavese N, Hotton G, et al. (2006) In vivo imaging of microglial activation with [11C](R)-PK11195 PET in idiopathic Parkinson's disease. Neurobiol Dis 21: 404–412. doi: 10.1016/j.nbd.2005.08.002
|
| [211] |
McGeer PL, Itagaki S, Boyes BE, et al. (1988) Reactive microglia are positive for HLA-DR in the substantia nigra of Parkinson's and Alzheimer's disease brains. Neurology 38: 1285–1291. doi: 10.1212/WNL.38.8.1285
|
| [212] | Nagatsu T, Mogi M, Ichinose H, et al. (2000) Changes in cytokines and neurotrophins in Parkinson's disease. J Neural Transm Suppl 80: 277–290. |
| [213] | Mengel D, Thelen M, Balzer-Geldsetzer M, et al. (2016) TREM2 rare variant p.R47H is not associated with Parkinson's disease. Parkinsonism Relat Disord 23: 109–111. |
| [214] |
Tay TL, Béchade C, D'Andrea I, et al. (2018) Microglia gone rogue: Impacts on psychiatric disorders across the lifespan. Front Mol Neurosci 10: 421. doi: 10.3389/fnmol.2017.00421
|
| [215] |
Pascual O, Ben Achour S, Rostaing P, et al. (2012) Microglia activation triggers astrocyte-mediated modulation of excitatory neurotransmission. Proc Natl Acad Sci U S A 109: E197–E205. doi: 10.1073/pnas.1111098109
|
| [216] |
Liddelow SA, Guttenplan KA, Clarke LE, et al. (2017) Neurotoxic reactive astrocytes are induced by activated microglia. Nature 541: 481–487. doi: 10.1038/nature21029
|
| [217] |
Goldmann T, Wieghofer P, Müller PF, et al. (2013) A new type of microglia gene targeting shows TAK1 to be pivotal in CNS autoimmune inflammation. Nat Neurosci 16: 1618–1626. doi: 10.1038/nn.3531
|
| [218] |
Yona S, Kim KW, Wolf Y, et al. (2013) Fate mapping reveals origins and dynamics of monocytes and tissue macrophages under homeostasis. Immunity 38: 79–91. doi: 10.1016/j.immuni.2012.12.001
|
| [219] |
Voronova A, Yuzwa SA, Wang BS, et al. (2017) Migrating interneurons secrete fractalkine to promote oligodendrocyte formation in the developing mammalian brain. Neuron 94: 500. doi: 10.1016/j.neuron.2017.04.018
|
| [220] |
Garré JM, Silva HM, Lafaille JJ, et al. (2017) CX3CR1+ monocytes modulate learning and learning-dependent dendritic spine remodeling via TNF-α. Nat Med 23: 714–722. doi: 10.1038/nm.4340
|
| [221] |
Goldmann T, Wieghofer P, Jord?o MJC, et al. (2016) Origin, fate and dynamics of macrophages at central nervous system interfaces. Nat Immunol 17: 797–805. doi: 10.1038/ni.3423
|
| [222] |
Buttgereit A, Lelios I, Yu X, et al. (2016) Sall1 is a transcriptional regulator defining microglia identity and function. Nat Immunol 17: 1397–1406. doi: 10.1038/ni.3585
|
| [223] |
Koso H, Tsuhako A, Lai CY, et al. (2016) Conditional rod photoreceptor ablation reveals Sall1 as a microglial marker and regulator of microglial morphology in the retina. Glia 64: 2005–2024. doi: 10.1002/glia.23038
|
| [224] | VanRyzin JW, Yu SJ, Perez-Pouchoulen M, et al. (2016) Temporary depletion of microglia during the early postnatal period induces lasting sex-dependent and sex-independent effects on behavior in rats. eNeuro 3: ENEURO.0297–16.2016. |
| [225] |
Grathwohl SA, K?lin RE, Bolmont T, et al. (2009) Formation and maintenance of Alzheimer's disease beta-amyloid plaques in the absence of microglia. Nat Neurosci 12: 1361–1363. doi: 10.1038/nn.2432
|
| [226] |
Dagher NN, Najafi AR, Kayala KMN, et al. (2015) Colony-stimulating factor 1 receptor inhibition prevents microglial plaque association and improves cognition in 3xTg-AD mice. J Neuroinflammation 12: 139. doi: 10.1186/s12974-015-0366-9
|
| [227] |
Spangenberg EE, Lee RJ, Najafi AR, et al. (2016) Eliminating microglia in Alzheimer's mice prevents neuronal loss without modulating amyloid-β pathology. Brain 139: 1265–1281. doi: 10.1093/brain/aww016
|
| [228] |
Olmos-Alonso A, Schetters STT, Sri S, et al. (2016) Pharmacological targeting of CSF1R inhibits microglial proliferation and prevents the progression of Alzheimer's-like pathology. Brain 139: 891–907. doi: 10.1093/brain/awv379
|
| [229] |
Asai H, Ikezu S, Tsunoda S, et al. (2015) Depletion of microglia and inhibition of exosome synthesis halt tau propagation. Nat Neurosci 18: 1584–1593. doi: 10.1038/nn.4132
|
| [230] |
Zhao R, Hu W, Tsai J, et al. (2017) Microglia limit the expansion of β-amyloid plaques in a mouse model of Alzheimer's disease. Mol Neurodegener 12: 47. doi: 10.1186/s13024-017-0188-6
|
| [231] |
Condello C, Yuan P, Schain A, et al. (2015) Microglia constitute a barrier that prevents neurotoxic protofibrillar Aβ42 hotspots around plaques. Nat Commun 6: 6176. doi: 10.1038/ncomms7176
|
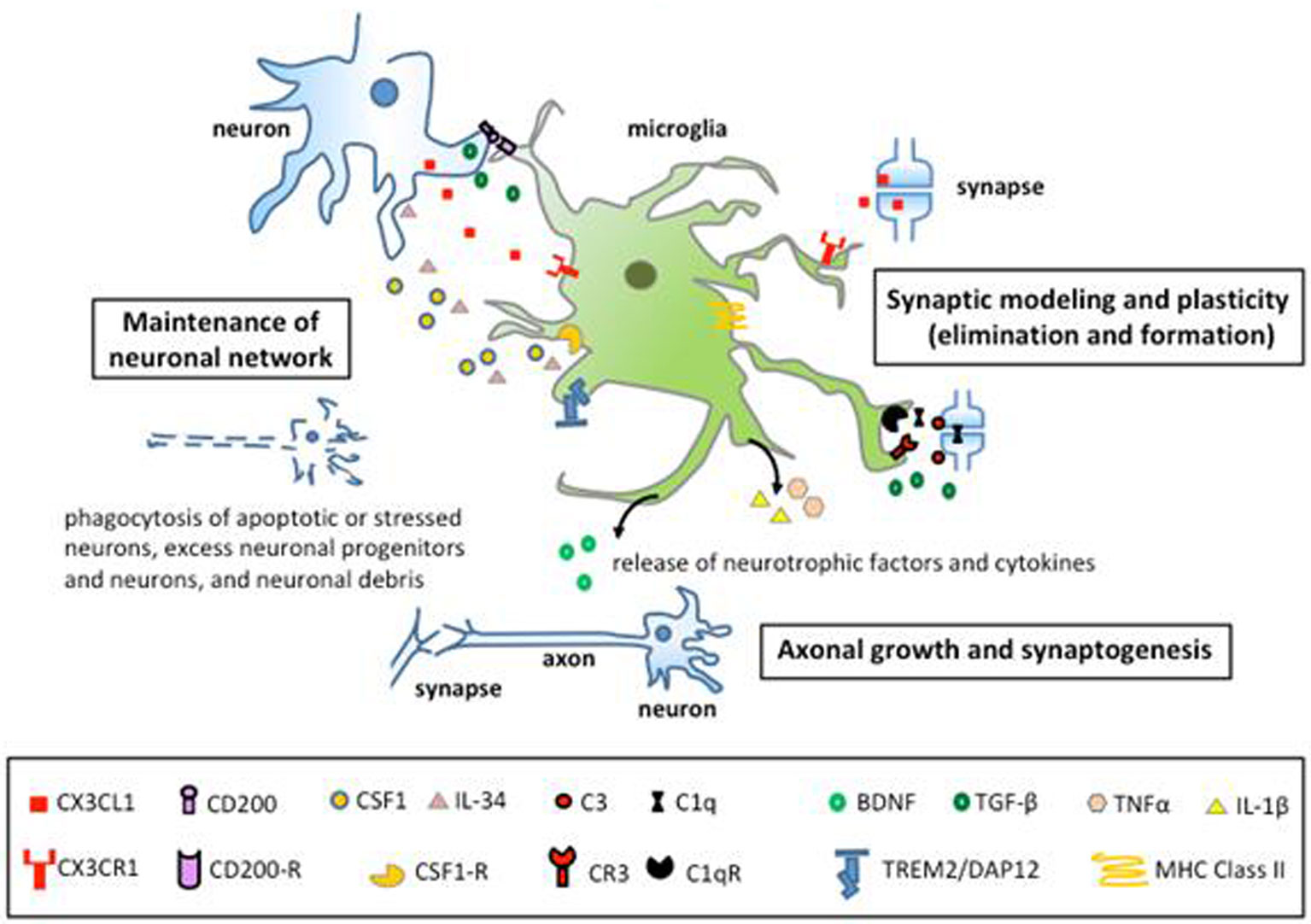
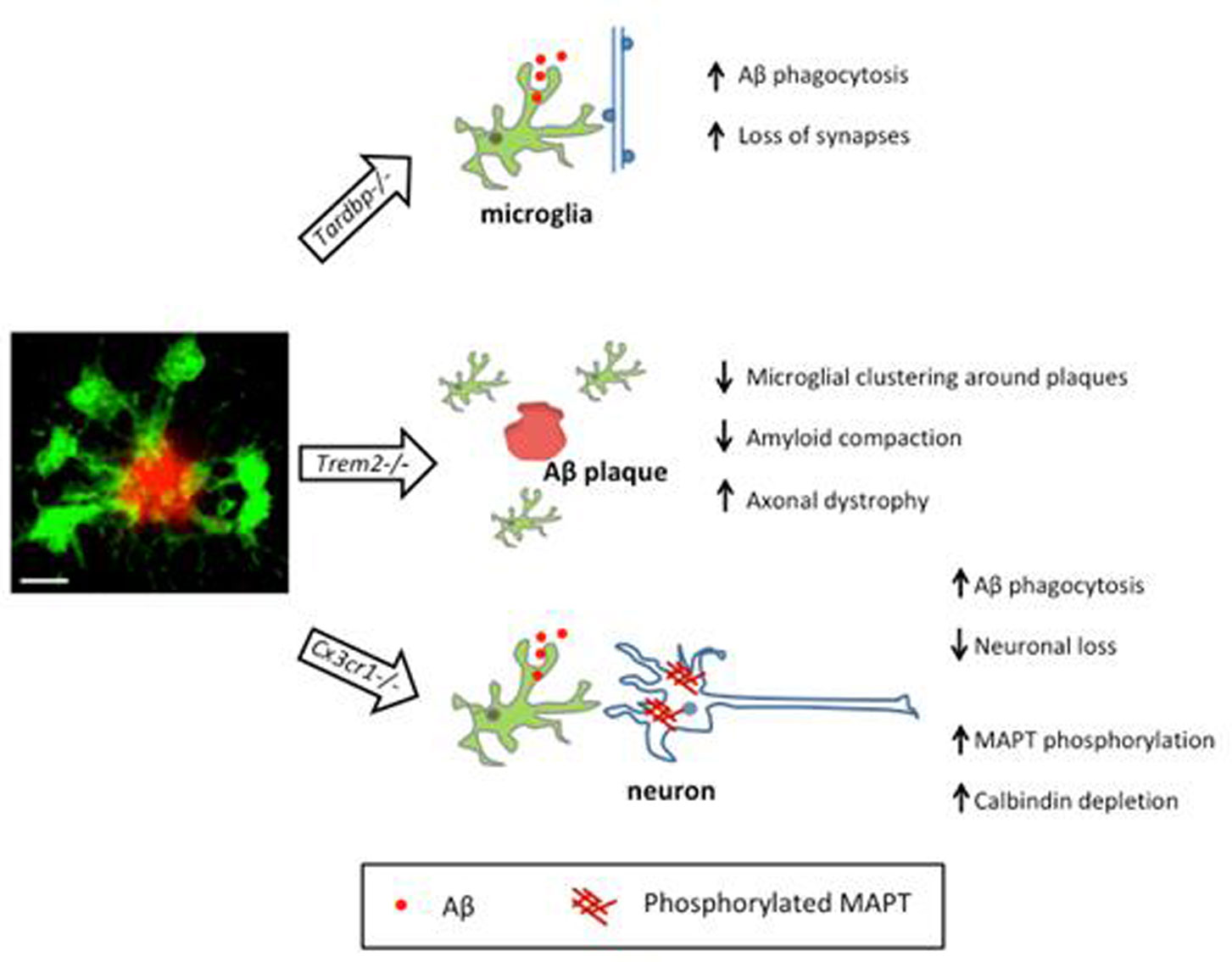
Figures(2)

Laetitia Weinhard, Paolo d'Errico, Tuan Leng Tay. Headmasters: Microglial regulation of learning and memory in health and disease[J]. AIMS Molecular Science, 2018, 5(1): 63-89. doi: 10.3934/molsci.2018.1.63







 DownLoad:
DownLoad: