Citation: Michael J. Murray. Drosophila models of metastasis[J]. AIMS Genetics, 2015, 2(1): 25-53. doi: 10.3934/genet.2015.1.25

| [1] |
Thiery JP, Acloque H, Huang RY, et al. (2009) Epithelial-mesenchymal transitions in development and disease. Cell 139: 871-890. doi: 10.1016/j.cell.2009.11.007

|
| [2] |
Chaffer CL, Thompson EW, Williams ED (2007) Mesenchymal to Epithelial Transition in Development and Disease. Cells Tissues Organs 185: 7-19. doi: 10.1159/000101298
|
| [3] |
Yao D, Dai C, Peng S (2011) Mechanism of the Mesenchymal-Epithelial Transition and Its Relationship with Metastatic Tumor Formation. Mol Cancer Res 9: 1608-1620. doi: 10.1158/1541-7786.MCR-10-0568
|
| [4] |
Friedl P, Wolf K (2003) Tumour-cell invasion and migration: diversity and escape mechanisms. Nat Rev Cancer 3: 362-374. doi: 10.1038/nrc1075
|
| [5] |
Calbo J, van Montfort E, Proost N, et al. (2011) A functional role for tumor cell heterogeneity in a mouse model of small cell lung cancer. Cancer Cell 19: 244-256. doi: 10.1016/j.ccr.2010.12.021
|
| [6] |
Mateo F, Meca-Cortes O, Celia-Terrassa T, et al. (2014) SPARC mediates metastatic cooperation between CSC and non-CSC prostate cancer cell subpopulations. Mol Cancer 13: 237. doi: 10.1186/1476-4598-13-237
|
| [7] |
Tsuji T, Ibaragi S, Hu GF (2009) Epithelial-mesenchymal transition and cell cooperativity in metastasis. Cancer Res 69: 7135-7139. doi: 10.1158/0008-5472.CAN-09-1618
|
| [8] |
Nakashima Y, Yoshinaga K, Kitao H, et al. (2013) Podoplanin is expressed at the invasive front of esophageal squamous cell carcinomas and is involved in collective cell invasion. Cancer Sci 104: 1718-1725. doi: 10.1111/cas.12286
|
| [9] |
Wicki A, Lehembre F, Wick N, et al. (2006) Tumor invasion in the absence of epithelial-mesenchymal transition: podoplanin-mediated remodeling of the actin cytoskeleton. Cancer Cell 9: 261-272. doi: 10.1016/j.ccr.2006.03.010
|
| [10] |
Nieto MA (2013) Epithelial plasticity: a common theme in embryonic and cancer cells. Science 342: 1234850. doi: 10.1126/science.1234850
|
| [11] |
Montell DJ (2006) The social lives of migrating cells in Drosophila. Curr Opin Genet Dev 16: 374-383. doi: 10.1016/j.gde.2006.06.010
|
| [12] |
Montell DJ, Yoon WH, Starz-Gaiano M (2012) Group choreography: mechanisms orchestrating the collective movement of border cells. Nat Rev Mol Cell Biol 13: 631-645. doi: 10.1038/nrm3433
|
| [13] |
Pocha SM, Montell DJ (2014) Cellular and Molecular Mechanisms of Single and Collective Cell Migrations in Drosophila: Themes and Variations. Annu Rev Genet 48: 295-318. doi: 10.1146/annurev-genet-120213-092218
|
| [14] |
Ribeiro C, Petit V, Affolter M (2003) Signaling systems, guided cell migration, and organogenesis: insights from genetic studies in Drosophila. Dev Biol 260: 1-8. doi: 10.1016/S0012-1606(03)00211-2
|
| [15] |
Wilson R, Leptin M (2000) Fibroblast growth factor receptor-dependent morphogenesis of the Drosophila mesoderm. Philos Trans R Soc Lond B Biol Sci 355: 891-895. doi: 10.1098/rstb.2000.0625
|
| [16] |
Winklbauer R, Muller HA (2011) Mesoderm layer formation in Xenopus and Drosophila gastrulation. Phys Biol 8: 045001. doi: 10.1088/1478-3975/8/4/045001
|
| [17] |
Nieto MA (2011) The ins and outs of the epithelial to mesenchymal transition in health and disease. Annu Rev Cell Dev Biol 27: 347-376. doi: 10.1146/annurev-cellbio-092910-154036
|
| [18] |
Affolter M, Caussinus E (2008) Tracheal branching morphogenesis in Drosophila: new insights into cell behaviour and organ architecture. Development 135: 2055-2064. doi: 10.1242/dev.014498
|
| [19] |
Harris TJ, Sawyer JK, Peifer M (2009) How the cytoskeleton helps build the embryonic body plan: models of morphogenesis from Drosophila. Curr Top Dev Biol 89: 55-85. doi: 10.1016/S0070-2153(09)89003-0
|
| [20] | Simpson P (1983) Maternal-Zygotic Gene Interactions during Formation of the Dorsoventral Pattern in Drosophila Embryos. Genetics 105: 615-632. |
| [21] |
Khan MA, Chen HC, Zhang D, et al. (2013) Twist: a molecular target in cancer therapeutics. Tumour Biol 34: 2497-2506. doi: 10.1007/s13277-013-1002-x
|
| [22] |
Wang Y, Shi J, Chai K, et al. (2013) The Role of Snail in EMT and Tumorigenesis. Curr Cancer Drug Targets 13: 963-972. doi: 10.2174/15680096113136660102
|
| [23] |
Sandmann T, Girardot C, Brehme M, et al. (2007) A core transcriptional network for early mesoderm development in Drosophila melanogaster. Genes Dev 21: 436-449. doi: 10.1101/gad.1509007
|
| [24] |
Leptin M (2005) Gastrulation movements: the logic and the nuts and bolts. Dev Cell 8: 305-320. doi: 10.1016/j.devcel.2005.02.007
|
| [25] |
Manning AJ, Rogers SL (2014) The Fog signaling pathway: insights into signaling in morphogenesis. Dev Biol 394: 6-14. doi: 10.1016/j.ydbio.2014.08.003
|
| [26] |
Murray MJ, Southall TD, Liu W, et al. (2012) Snail-dependent repression of the RhoGEF pebble is required for gastrulation consistency in Drosophila melanogaster. Dev Genes Evol 222: 361-368. doi: 10.1007/s00427-012-0414-8
|
| [27] |
Wheelock MJ, Shintani Y, Maeda M, et al. (2008) Cadherin switching. J Cell Sci 121: 727-735. doi: 10.1242/jcs.000455
|
| [28] |
Jeanes A, Gottardi CJ, Yap AS (2008) Cadherins and cancer: how does cadherin dysfunction promote tumor progression? Oncogene 27: 6920-6929. doi: 10.1038/onc.2008.343
|
| [29] |
Mariotti A, Perotti A, Sessa C, et al. (2007) N-cadherin as a therapeutic target in cancer. Expert Opin Investig Drugs 16: 451-465. doi: 10.1517/13543784.16.4.451
|
| [30] |
Oda H, Tsukita S, Takeichi M (1998) Dynamic behavior of the cadherin-based cell-cell adhesion system during Drosophila gastrulation. Dev Biol 203: 435-450. doi: 10.1006/dbio.1998.9047
|
| [31] |
Schafer G, Narasimha M, Vogelsang E, et al. (2014) Cadherin switching during the formation and differentiation of the Drosophila mesoderm - implications for epithelial-to-mesenchymal transitions. J Cell Sci 127: 1511-1522. doi: 10.1242/jcs.139485
|
| [32] |
Clark IB, Muha V, Klingseisen A, et al. (2011) Fibroblast growth factor signalling controls successive cell behaviours during mesoderm layer formation in Drosophila. Development 138: 2705-2715. doi: 10.1242/dev.060277
|
| [33] |
Murray MJ, Saint R (2007) Photoactivatable GFP resolves Drosophila mesoderm migration behaviour. Development 134: 3975-3983. doi: 10.1242/dev.005389
|
| [34] |
Smallhorn M, Murray MJ, Saint R (2004) The epithelial-mesenchymal transition of the Drosophila mesoderm requires the Rho GTP exchange factor Pebble. Development 131: 2641-2651. doi: 10.1242/dev.01150
|
| [35] |
Williams M, Burdsal C, Periasamy A, et al. (2012) Mouse primitive streak forms in situ by initiation of epithelial to mesenchymal transition without migration of a cell population. Dev Dyn 241: 270-283. doi: 10.1002/dvdy.23711
|
| [36] |
Berndt JD, Clay MR, Langenberg T, et al. (2008) Rho-kinase and myosin II affect dynamic neural crest cell behaviors during epithelial to mesenchymal transition in vivo. Dev Biol 324: 236-244. doi: 10.1016/j.ydbio.2008.09.013
|
| [37] |
Eisenhoffer GT, Rosenblatt J (2013) Bringing balance by force: live cell extrusion controls epithelial cell numbers. Trends Cell Biol 23: 185-192. doi: 10.1016/j.tcb.2012.11.006
|
| [38] | Polakis P (2012) Wnt signaling in cancer. Cold Spring Harb Perspect Biol 4. |
| [39] |
Rembold M, Ciglar L, Yanez-Cuna JO, et al. (2014) A conserved role for Snail as a potentiator of active transcription. Genes Dev 28: 167-181. doi: 10.1101/gad.230953.113
|
| [40] |
Hahn S, Jackstadt R, Siemens H, et al. (2013) SNAIL and miR-34a feed-forward regulation of ZNF281/ZBP99 promotes epithelial-mesenchymal transition. EMBO J 32: 3079-3095. doi: 10.1038/emboj.2013.236
|
| [41] |
Stemmer V, de Craene B, Berx G, et al. (2008) Snail promotes Wnt target gene expression and interacts with beta-catenin. Oncogene 27: 5075-5080. doi: 10.1038/onc.2008.140
|
| [42] |
Wels C, Joshi S, Koefinger P, et al. (2011) Transcriptional activation of ZEB1 by Slug leads to cooperative regulation of the epithelial-mesenchymal transition-like phenotype in melanoma. J Invest Dermatol 131: 1877-1885. doi: 10.1038/jid.2011.142
|
| [43] |
Tao G, Levay AK, Gridley T, et al. (2011) Mmp15 is a direct target of Snai1 during endothelial to mesenchymal transformation and endocardial cushion development. Dev Biol 359: 209-221. doi: 10.1016/j.ydbio.2011.08.022
|
| [44] |
Campbell K, Whissell G, Franch-Marro X, et al. (2011) Specific GATA factors act as conserved inducers of an endodermal-EMT. Dev Cell 21: 1051-1061. doi: 10.1016/j.devcel.2011.10.005
|
| [45] |
Godde NJ, Pearson HB, Smith LK, et al. (2014) Dissecting the role of polarity regulators in cancer through the use of mouse models. Exp Cell Res 328: 249-257. doi: 10.1016/j.yexcr.2014.08.036
|
| [46] |
Humbert PO, Grzeschik NA, Brumby AM, et al. (2008) Control of tumourigenesis by the Scribble/Dlg/Lgl polarity module. Oncogene 27: 6888-6907. doi: 10.1038/onc.2008.341
|
| [47] |
Khursheed M, Bashyam MD (2014) Apico-basal polarity complex and cancer. J Biosci 39: 145-155. doi: 10.1007/s12038-013-9410-z
|
| [48] | Gao X, Sedgwick T, Shi YB, et al. (1998) Distinct functions are implicated for the GATA-4, -5, and -6 transcription factors in the regulation of intestine epithelial cell differentiation. Mol Cell Biol 18: 2901-2911. |
| [49] | Molkentin JD, Antos C, Mercer B, et al. (2000) Direct activation of a GATA6 cardiac enhancer by Nkx2.5: evidence for a reinforcing regulatory network of Nkx2.5 and GATA transcription factors in the developing heart. Dev Biol 217: 301-309. |
| [50] |
Fu B, Luo M, Lakkur S, et al. (2008) Frequent genomic copy number gain and overexpression of GATA-6 in pancreatic carcinoma. Cancer Biol Ther 7: 1593-1601. doi: 10.4161/cbt.7.10.6565
|
| [51] |
Kwei KA, Bashyam MD, Kao J, et al. (2008) Genomic profiling identifies GATA6 as a candidate oncogene amplified in pancreatobiliary cancer. PLoS Genet 4: e1000081. doi: 10.1371/journal.pgen.1000081
|
| [52] |
Shureiqi I, Zuo X, Broaddus R, et al. (2007) The transcription factor GATA-6 is overexpressed in vivo and contributes to silencing 15-LOX-1 in vitro in human colon cancer. FASEB J 21: 743-753. doi: 10.1096/fj.06-6830com
|
| [53] |
Bokel C, Brown NH (2002) Integrins in development: moving on, responding to, and sticking to the extracellular matrix. Dev Cell 3: 311-321. doi: 10.1016/S1534-5807(02)00265-4
|
| [54] |
Devenport D, Brown NH (2004) Morphogenesis in the absence of integrins: mutation of both Drosophila beta subunits prevents midgut migration. Development 131: 5405-5415. doi: 10.1242/dev.01427
|
| [55] | Martin-Bermudo MD, Alvarez-Garcia I, Brown NH (1999) Migration of the Drosophila primordial midgut cells requires coordination of diverse PS integrin functions. Development 126: 5161-5169. |
| [56] |
Urbano JM, Dominguez-Gimenez P, Estrada B, et al. (2011) PS integrins and laminins: key regulators of cell migration during Drosophila embryogenesis. PLoS One 6: e23893. doi: 10.1371/journal.pone.0023893
|
| [57] | Pert M, Gan M, Saint R, et al. (2015) Netrins and Frazzled/DCC promote the migration and mesenchymal to epithelial transition of Drosophila midgut cells. Biology Open. doi:10.1242/bio.201410827 [Epub ahead of print] |
| [58] |
Tepass U, Hartenstein V (1994) The development of cellular junctions in the Drosophila embryo. Dev Biol 161: 563-596. doi: 10.1006/dbio.1994.1054
|
| [59] | Tepass U, Hartenstein V (1994) Epithelium formation in the Drosophila midgut depends on the interaction of endoderm and mesoderm. Development 120: 579-590. |
| [60] |
Yarnitzky T, Volk T (1995) Laminin is required for heart, somatic muscles, and gut development in the Drosophila embryo. Dev Biol 169: 609-618. doi: 10.1006/dbio.1995.1173
|
| [61] |
Pastor-Pareja JC, Grawe F, Martin-Blanco E, et al. (2004) Invasive cell behavior during Drosophila imaginal disc eversion is mediated by the JNK signaling cascade. Dev Cell 7: 387-399. doi: 10.1016/j.devcel.2004.07.022
|
| [62] |
Martin-Blanco E, Pastor-Pareja JC, Garcia-Bellido A (2000) JNK and decapentaplegic signaling control adhesiveness and cytoskeleton dynamics during thorax closure in Drosophila. Proc Natl Acad Sci U S A 97: 7888-7893. doi: 10.1073/pnas.97.14.7888
|
| [63] |
Tateno M, Nishida Y, Adachi-Yamada T (2000) Regulation of JNK by Src during Drosophila development. Science 287: 324-327. doi: 10.1126/science.287.5451.324
|
| [64] |
Ishimaru S, Ueda R, Hinohara Y, et al. (2004) PVR plays a critical role via JNK activation in thorax closure during Drosophila metamorphosis. EMBO J 23: 3984-3994. doi: 10.1038/sj.emboj.7600417
|
| [65] |
Srivastava A, Pastor-Pareja JC, Igaki T, et al. (2007) Basement membrane remodeling is essential for Drosophila disc eversion and tumor invasion. Proc Natl Acad Sci U S A 104: 2721-2726. doi: 10.1073/pnas.0611666104
|
| [66] |
Homsy JG, Jasper H, Peralta XG, et al. (2006) JNK signaling coordinates integrin and actin functions during Drosophila embryogenesis. Dev Dyn 235: 427-434. doi: 10.1002/dvdy.20649
|
| [67] |
Bubici C, Papa S (2014) JNK signalling in cancer: in need of new, smarter therapeutic targets. Br J Pharmacol 171: 24-37. doi: 10.1111/bph.12432
|
| [68] | Manhire-Heath R, Golenkina S, Saint R, et al. (2013) Netrin-dependent downregulation of Frazzled/DCC is required for the dissociation of the peripodial epithelium in Drosophila. Nat Commun 4: 2790. |
| [69] |
Speck O, Hughes SC, Noren NK, et al. (2003) Moesin functions antagonistically to the Rho pathway to maintain epithelial integrity. Nature 421: 83-87. doi: 10.1038/nature01295
|
| [70] |
VanZomeren-Dohm A, Sarro J, Flannery E, et al. (2011) The Drosophila Netrin receptor frazzled/DCC functions as an invasive tumor suppressor. BMC Dev Biol 11: 41. doi: 10.1186/1471-213X-11-41
|
| [71] | Castets M, Broutier L, Molin Y, et al. (2012) DCC constrains tumour progression via its dependence receptor activity. Nature 482: 534-537. |
| [72] |
Fitamant J, Guenebeaud C, Coissieux MM, et al. (2008) Netrin-1 expression confers a selective advantage for tumor cell survival in metastatic breast cancer. Proc Natl Acad Sci U S A 105: 4850-4855. doi: 10.1073/pnas.0709810105
|
| [73] | Papanastasiou AD, Pampalakis G, Katsaros D, et al. (2011) Netrin-1 overexpression is predictive of ovarian malignancies. Oncotarget 2: 363-367. |
| [74] |
Mazelin L, Bernet A, Bonod-Bidaud C, et al. (2004) Netrin-1 controls colorectal tumorigenesis by regulating apoptosis. Nature 431: 80-84. doi: 10.1038/nature02788
|
| [75] |
Lai Wing Sun K, Correia JP, Kennedy TE (2011) Netrins: versatile extracellular cues with diverse functions. Development 138: 2153-2169. doi: 10.1242/dev.044529
|
| [76] |
Dumartin L, Quemener C, Laklai H, et al. (2010) Netrin-1 mediates early events in pancreatic adenocarcinoma progression, acting on tumor and endothelial cells. Gastroenterology 138: 1595-1606, 1606 e1591-1598. doi: 10.1053/j.gastro.2009.12.061
|
| [77] | Kaufmann S, Kuphal S, Schubert T, et al. (2009) Functional implication of Netrin expression in malignant melanoma. Cell Oncol 31: 415-422. |
| [78] |
Rodrigues S, De Wever O, Bruyneel E, et al. (2007) Opposing roles of netrin-1 and the dependence receptor DCC in cancer cell invasion, tumor growth and metastasis. Oncogene 26: 5615-5625. doi: 10.1038/sj.onc.1210347
|
| [79] | Salminen M, Meyer BI, Bober E, et al. (2000) Netrin 1 is required for semicircular canal formation in the mouse inner ear. Development 127: 13-22. |
| [80] |
Martin M, Simon-Assmann P, Kedinger M, et al. (2006) DCC regulates cell adhesion in human colon cancer derived HT-29 cells and associates with ezrin. Eur J Cell Biol 85: 769-783. doi: 10.1016/j.ejcb.2006.02.013
|
| [81] |
Kee N, Wilson N, De Vries M, et al. (2008) Neogenin and RGMa control neural tube closure and neuroepithelial morphology by regulating cell polarity. J Neurosci 28: 12643-12653. doi: 10.1523/JNEUROSCI.4265-08.2008
|
| [82] |
Fulga TA, Rorth P (2002) Invasive cell migration is initiated by guided growth of long cellular extensions. Nat Cell Biol 4: 715-719. doi: 10.1038/ncb848
|
| [83] |
Majumder P, Aranjuez G, Amick J, et al. (2012) Par-1 controls myosin-II activity through myosin phosphatase to regulate border cell migration. Curr Biol 22: 363-372. doi: 10.1016/j.cub.2012.01.037
|
| [84] |
Wang X, Adam JC, Montell D (2007) Spatially localized Kuzbanian required for specific activation of Notch during border cell migration. Dev Biol 301: 532-540. doi: 10.1016/j.ydbio.2006.08.031
|
| [85] |
Prasad M, Montell DJ (2007) Cellular and molecular mechanisms of border cell migration analyzed using time-lapse live-cell imaging. Dev Cell 12: 997-1005. doi: 10.1016/j.devcel.2007.03.021
|
| [86] |
Ara T, Declerck YA (2010) Interleukin-6 in bone metastasis and cancer progression. Eur J Cancer 46: 1223-1231. doi: 10.1016/j.ejca.2010.02.026
|
| [87] |
Sullivan NJ, Sasser AK, Axel AE, et al. (2009) Interleukin-6 induces an epithelial-mesenchymal transition phenotype in human breast cancer cells. Oncogene 28: 2940-2947. doi: 10.1038/onc.2009.180
|
| [88] |
Min Y, Ghose S, Boelte K, et al. (2011) C/EBP-delta regulates VEGF-C autocrine signaling in lymphangiogenesis and metastasis of lung cancer through HIF-1alpha. Oncogene 30: 4901-4909. doi: 10.1038/onc.2011.187
|
| [89] |
Doronkin S, Djagaeva I, Nagle ME, et al. (2010) Dose-dependent modulation of HIF-1alpha/sima controls the rate of cell migration and invasion in Drosophila ovary border cells. Oncogene 29: 1123-1134. doi: 10.1038/onc.2009.407
|
| [90] |
Anzick SL, Kononen J, Walker RL, et al. (1997) AIB1, a steroid receptor coactivator amplified in breast and ovarian cancer. Science 277: 965-968. doi: 10.1126/science.277.5328.965
|
| [91] |
Mazzone M, Selfors LM, Albeck J, et al. (2010) Dose-dependent induction of distinct phenotypic responses to Notch pathway activation in mammary epithelial cells. Proc Natl Acad Sci U S A 107: 5012-5017. doi: 10.1073/pnas.1000896107
|
| [92] |
Bell GP, Thompson BJ (2014) Colorectal cancer progression: lessons from Drosophila? Semin Cell Dev Biol 28: 70-77. doi: 10.1016/j.semcdb.2014.02.007
|
| [93] |
Gandille P, Narbonne-Reveau K, Boissonneau E, et al. (2010) Mutations in the polycomb group gene polyhomeotic lead to epithelial instability in both the ovary and wing imaginal disc in Drosophila. PLoS One 5: e13946. doi: 10.1371/journal.pone.0013946
|
| [94] |
Widmann TJ, Dahmann C (2009) Dpp signaling promotes the cuboidal-to-columnar shape transition of Drosophila wing disc epithelia by regulating Rho1. J Cell Sci 122: 1362-1373. doi: 10.1242/jcs.044271
|
| [95] |
Marinari E, Mehonic A, Curran S, et al. (2012) Live-cell delamination counterbalances epithelial growth to limit tissue overcrowding. Nature 484: 542-545. doi: 10.1038/nature10984
|
| [96] |
Tepass U (2012) The apical polarity protein network in Drosophila epithelial cells: regulation of polarity, junctions, morphogenesis, cell growth, and survival. Annu Rev Cell Dev Biol 28: 655-685. doi: 10.1146/annurev-cellbio-092910-154033
|
| [97] |
Bilder D, Li M, Perrimon N (2000) Cooperative regulation of cell polarity and growth by Drosophila tumor suppressors. Science 289: 113-116. doi: 10.1126/science.289.5476.113
|
| [98] |
Brumby AM, Goulding KR, Schlosser T, et al. (2011) Identification of novel Ras-cooperating oncogenes in Drosophila melanogaster: a RhoGEF/Rho-family/JNK pathway is a central driver of tumorigenesis. Genetics 188: 105-125. doi: 10.1534/genetics.111.127910
|
| [99] |
Ohsawa S, Sugimura K, Takino K, et al. (2011) Elimination of oncogenic neighbors by JNK-mediated engulfment in Drosophila. Dev Cell 20: 315-328. doi: 10.1016/j.devcel.2011.02.007
|
| [100] |
Uhlirova M, Bohmann D (2006) JNK- and Fos-regulated Mmp1 expression cooperates with Ras to induce invasive tumors in Drosophila. EMBO J 25: 5294-5304. doi: 10.1038/sj.emboj.7601401
|
| [101] |
Kulshammer E, Uhlirova M (2013) The actin cross-linker Filamin/Cheerio mediates tumor malignancy downstream of JNK signaling. J Cell Sci 126: 927-938. doi: 10.1242/jcs.114462
|
| [102] |
Brock AR, Wang Y, Berger S, et al. (2012) Transcriptional regulation of Profilin during wound closure in Drosophila larvae. J Cell Sci 125: 5667-5676. doi: 10.1242/jcs.107490
|
| [103] |
Dekanty A, Barrio L, Muzzopappa M, et al. (2012) Aneuploidy-induced delaminating cells drive tumorigenesis in Drosophila epithelia. Proc Natl Acad Sci U S A 109: 20549-20554. doi: 10.1073/pnas.1206675109
|
| [104] |
Hipfner DR, Keller N, Cohen SM (2004) Slik Sterile-20 kinase regulates Moesin activity to promote epithelial integrity during tissue growth. Genes Dev 18: 2243-2248. doi: 10.1101/gad.303304
|
| [105] |
Neisch AL, Formstecher E, Fehon RG (2013) Conundrum, an ARHGAP18 orthologue, regulates RhoA and proliferation through interactions with Moesin. Mol Biol Cell 24: 1420-1433. doi: 10.1091/mbc.E12-11-0800
|
| [106] |
Coso OA, Chiariello M, Yu JC, et al. (1995) The small GTP-binding proteins Rac1 and Cdc42 regulate the activity of the JNK/SAPK signaling pathway. Cell 81: 1137-1146. doi: 10.1016/S0092-8674(05)80018-2
|
| [107] | Teramoto H, Crespo P, Coso OA, et al. (1996) The small GTP-binding protein rho activates c-Jun N-terminal kinases/stress-activated protein kinases in human kidney 293T cells. Evidence for a Pak-independent signaling pathway. J Biol Chem 271: 25731-25734. |
| [108] |
Neisch AL, Speck O, Stronach B, et al. (2010) Rho1 regulates apoptosis via activation of the JNK signaling pathway at the plasma membrane. J Cell Biol 189: 311-323. doi: 10.1083/jcb.200912010
|
| [109] |
Khoo P, Allan K, Willoughby L, et al. (2013) In Drosophila, RhoGEF2 cooperates with activated Ras in tumorigenesis through a pathway involving Rho1-Rok-Myosin-II and JNK signalling. Dis Model Mech 6: 661-678. doi: 10.1242/dmm.010066
|
| [110] |
Vidal M, Larson DE, Cagan RL (2006) Csk-deficient boundary cells are eliminated from normal Drosophila epithelia by exclusion, migration, and apoptosis. Dev Cell 10: 33-44. doi: 10.1016/j.devcel.2005.11.007
|
| [111] |
Rudrapatna VA, Bangi E, Cagan RL (2014) A Jnk-Rho-Actin remodeling positive feedback network directs Src-driven invasion. Oncogene 33: 2801-2806. doi: 10.1038/onc.2013.232
|
| [112] |
Singh J, Aaronson SA, Mlodzik M (2010) Drosophila Abelson kinase mediates cell invasion and proliferation through two distinct MAPK pathways. Oncogene 29: 4033-4045. doi: 10.1038/onc.2010.155
|
| [113] |
Das TK, Sangodkar J, Negre N, et al. (2013) Sin3a acts through a multi-gene module to regulate invasion in Drosophila and human tumors. Oncogene 32: 3184-3197. doi: 10.1038/onc.2012.326
|
| [114] |
Brumby AM, Richardson HE (2003) scribble mutants cooperate with oncogenic Ras or Notch to cause neoplastic overgrowth in Drosophila. EMBO J 22: 5769-5779. doi: 10.1093/emboj/cdg548
|
| [115] |
Pagliarini RA, Xu T (2003) A genetic screen in Drosophila for metastatic behavior. Science 302: 1227-1231. doi: 10.1126/science.1088474
|
| [116] |
Igaki T, Pagliarini RA, Xu T (2006) Loss of cell polarity drives tumor growth and invasion through JNK activation in Drosophila. Curr Biol 16: 1139-1146. doi: 10.1016/j.cub.2006.04.042
|
| [117] |
Ma X, Shao Y, Zheng H, et al. (2013) Src42A modulates tumor invasion and cell death via Ben/dUev1a-mediated JNK activation in Drosophila. Cell Death Dis 4: e864. doi: 10.1038/cddis.2013.392
|
| [118] |
Rudrapatna VA, Bangi E, Cagan RL (2013) Caspase signalling in the absence of apoptosis drives Jnk-dependent invasion. EMBO Rep 14: 172-177. doi: 10.1038/embor.2012.217
|
| [119] | Yang L, Cao Z, Yan H, et al. (2003) Coexistence of high levels of apoptotic signaling and inhibitor of apoptosis proteins in human tumor cells: implication for cancer specific therapy. Cancer Res 63: 6815-6824. |
| [120] |
Wu M, Pastor-Pareja JC, Xu T (2010) Interaction between Ras(V12) and scribbled clones induces tumour growth and invasion. Nature 463: 545-548. doi: 10.1038/nature08702
|
| [121] |
Herranz H, Hong X, Hung NT, et al. (2012) Oncogenic cooperation between SOCS family proteins and EGFR identified using a Drosophila epithelial transformation model. Genes Dev 26: 1602-1611. doi: 10.1101/gad.192021.112
|
| [122] |
de Visser KE, Eichten A, Coussens LM (2006) Paradoxical roles of the immune system during cancer development. Nat Rev Cancer 6: 24-37. doi: 10.1038/nrc1782
|
| [123] |
Hanahan D, Weinberg RA (2011) Hallmarks of cancer: the next generation. Cell 144: 646-674. doi: 10.1016/j.cell.2011.02.013
|
| [124] | Landskron G, De la Fuente M, Thuwajit P, et al. (2014) Chronic inflammation and cytokines in the tumor microenvironment. J Immunol Res 2014: 149185. |
| [125] |
Pastor-Pareja JC, Wu M, Xu T (2008) An innate immune response of blood cells to tumors and tissue damage in Drosophila. Dis Model Mech 1: 144-154; discussion 153. doi: 10.1242/dmm.000950
|
| [126] |
Cordero JB, Macagno JP, Stefanatos RK, et al. (2010) Oncogenic Ras diverts a host TNF tumor suppressor activity into tumor promoter. Dev Cell 18: 999-1011. doi: 10.1016/j.devcel.2010.05.014
|
| [127] | Shen J, Lu J, Sui L, et al. (2014) The orthologous Tbx transcription factors Omb and TBX2 induce epithelial cell migration and extrusion in vivo without involvement of matrix metalloproteinases. Oncotarget 5: 11998-12015. |
| [128] |
Das TK, Dana D, Paroly SS, et al. (2013) Centrosomal kinase Nek2 cooperates with oncogenic pathways to promote metastasis. Oncogenesis 2: e69. doi: 10.1038/oncsis.2013.34
|
| [129] |
Dominguez M (2014) Oncogenic programmes and Notch activity: an 'organized crime'? Semin Cell Dev Biol 28: 78-85. doi: 10.1016/j.semcdb.2014.04.012
|
| [130] |
Gratz SJ, Wildonger J, Harrison MM, et al. (2013) CRISPR/Cas9-mediated genome engineering and the promise of designer flies on demand. Fly (Austin) 7: 249-255. doi: 10.4161/fly.26566
|
| [131] | Kvon EZ, Kazmar T, Stampfel G, et al. (2014) Genome-scale functional characterization of Drosophila developmental enhancers in vivo. Nature 512: 91-95. |
| [132] |
Southall TD, Gold KS, Egger B, et al. (2013) Cell-type-specific profiling of gene expression and chromatin binding without cell isolation: assaying RNA Pol II occupancy in neural stem cells. Dev Cell 26: 101-112. doi: 10.1016/j.devcel.2013.05.020
|
| [133] |
Ferres-Marco D, Gutierrez-Garcia I, Vallejo DM, et al. (2006) Epigenetic silencers and Notch collaborate to promote malignant tumours by Rb silencing. Nature 439: 430-436. doi: 10.1038/nature04376
|
| [134] |
Willoughby LF, Schlosser T, Manning SA, et al. (2013) An in vivo large-scale chemical screening platform using Drosophila for anti-cancer drug discovery. Dis Model Mech 6: 521-529. doi: 10.1242/dmm.009985
|
| [135] |
Vidal M, Wells S, Ryan A, et al. (2005) ZD6474 suppresses oncogenic RET isoforms in a Drosophila model for type 2 multiple endocrine neoplasia syndromes and papillary thyroid carcinoma. Cancer Res 65: 3538-3541. doi: 10.1158/0008-5472.CAN-04-4561
|
| [136] |
Dar AC, Das TK, Shokat KM, et al. (2012) Chemical genetic discovery of targets and anti-targets for cancer polypharmacology. Nature 486: 80-84. doi: 10.1038/nature11127
|
| [137] |
Kasai Y, Cagan R (2010) Drosophila as a tool for personalized medicine: a primer. Per Med 7: 621-632. doi: 10.2217/pme.10.65
|
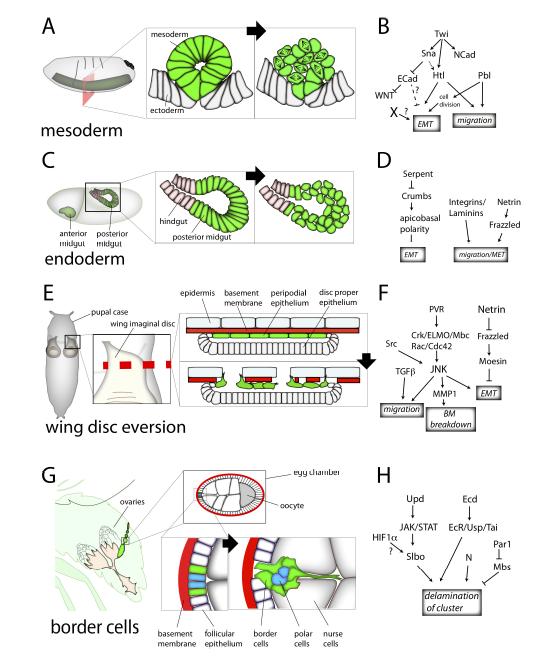
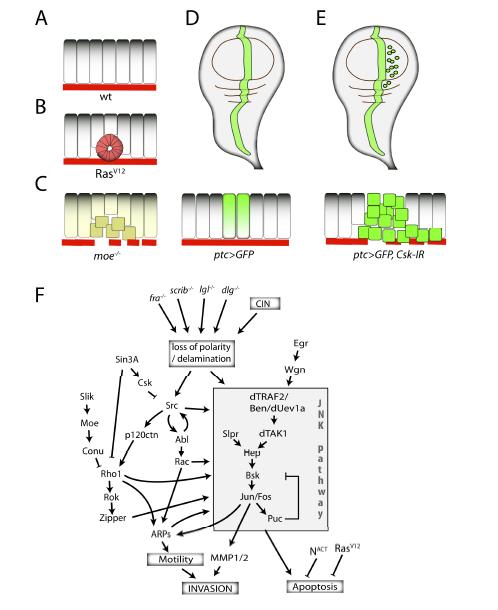
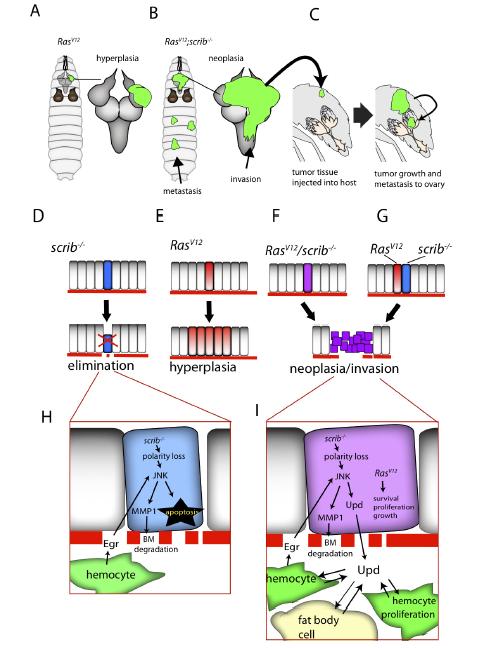
Figures(3) / Tables(1)

Michael J. Murray. Drosophila models of metastasis[J]. AIMS Genetics, 2015, 2(1): 25-53. doi: 10.3934/genet.2015.1.25







 DownLoad:
DownLoad: