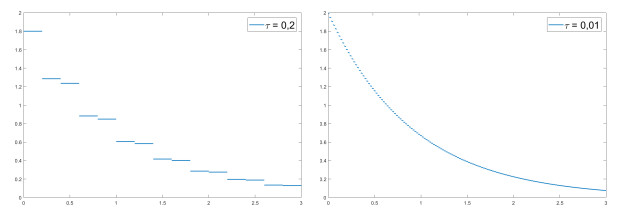
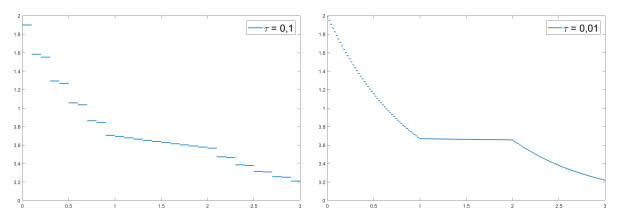
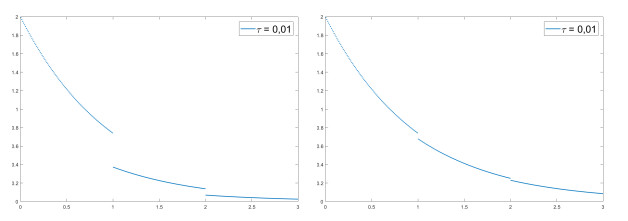
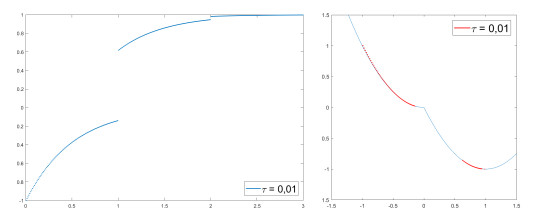
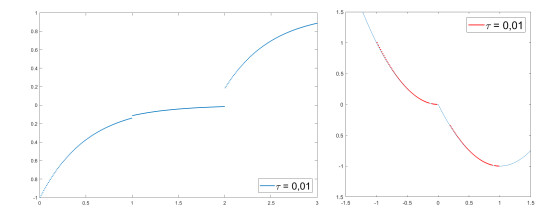
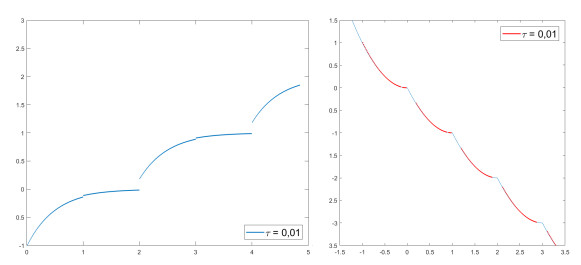
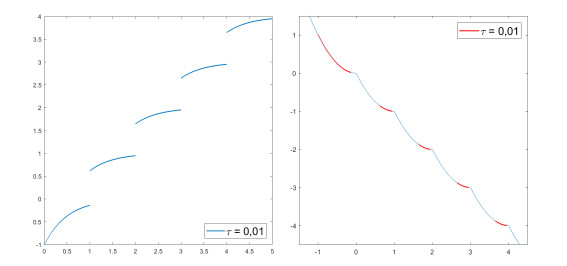
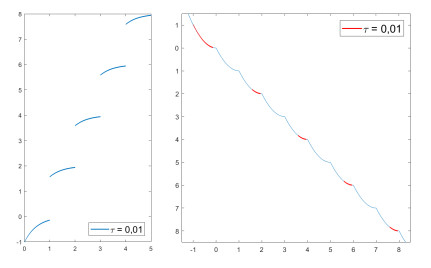
We modify the De Giorgi's minimizing movements scheme for a functional $φ$, by perturbing the dissipation term, and find a condition on the perturbations which ensures the convergence of the scheme to an absolutely continuous perturbed minimizing movement. The perturbations produce a variation of the metric derivative of the minimizing movement. This process is formalized by the introduction of the notion of curve of maximal slope for $φ$ with a given rate. We show that if we relax the condition on the perturbations we may have many different meaningful effects; in particular, some perturbed minimizing movements may explore different potential wells.
Citation: Antonio Tribuzio. Perturbations of minimizing movements and curves of maximal slope[J]. Networks and Heterogeneous Media, 2018, 13(3): 423-448. doi: 10.3934/nhm.2018019
We modify the De Giorgi's minimizing movements scheme for a functional $φ$, by perturbing the dissipation term, and find a condition on the perturbations which ensures the convergence of the scheme to an absolutely continuous perturbed minimizing movement. The perturbations produce a variation of the metric derivative of the minimizing movement. This process is formalized by the introduction of the notion of curve of maximal slope for $φ$ with a given rate. We show that if we relax the condition on the perturbations we may have many different meaningful effects; in particular, some perturbed minimizing movements may explore different potential wells.

| [1] |
N. Ansini, Gradient flows with wiggly potential: a variational approach to the dynamics, in
Mathematical Analysis of Continuum Mechanics and Industrial Applications Ⅱ, CoMFoS16
(Springer), 30 (2017), 139-151. doi: 10.1007/978-981-10-6283-4_12

|
| [2] | N. Ansini, A. Braides and J. Zimmer, Minimising movements for oscillating energies: The critical regime, Proceedings of the Royal Society of Edinburgh Section A (in press), 2016, arXiv: 1605.01885. |
| [3] |
Curvature-driven flows: a variational approach. SIAM Journal of Control and Optimization (1993) 31: 387-438.
|
| [4] | L. Ambrosio, N. Gigli and G. Savaré, Gradient Flows in Metric Spaces and in the Space of Probability Measures, Birkhauser Verlag, second edition, 2008. |
| [5] |
A. Braides,
Γ-convergence for Beginners, Oxford University Press, 2002. doi: 10.1093/acprof:oso/9780198507840.001.0001
|
| [6] |
A. Braides,
Local Minimization, Variational Evolution and Γ-convergence, Springer Cham, 2014. doi: 10.1007/978-3-319-01982-6
|
| [7] |
Minimizing movements along a sequence of functionals and curves of maximal slope. Comptes Rendus Matematique (2005) 354: 685-689.
|
| [8] |
M. Colombo and M. Gobbino, Passing to the limit in maximal slope curves: from a regularized Perona-Malik to the total variation flow,
Mathematical Models and Methods in Applied Sciences, 22 (2012), 1250017, 19pp. doi: 10.1142/S0218202512500170
|
| [9] |
L. C. Evans,
Partial Differential Equations, American Mathematical Society, second edition, 2010. doi: 10.1090/gsm/019
|
| [10] | F. Fleissner, Γ-convergence and relaxations for gradient flows in metric spaces: A minimizing movement approach, ESAIM Control, Optimisation and Calculus of Variations (to appear), preprint, arXiv: 1603.02822, (2016). |
| [11] |
F. Fleissner and G. Savaré, Reverse approximation of gradient flows as minimizing movements: A conjecture by De Giorgi,
Forthcoming Articles, (2018), 30pp, arXiv: 1711.07256. doi: 10.2422/2036-2145.201711_008
|
| [12] |
The variational formulation of the Fokker-Plank equation. SIAM Journal of Mathematical Analysis (1998) 29: 1-17.
|
| [13] |
Gamma-convergence of gradient flows and application to Ginzburg-Landau. Communications on Pure and Applied Mathematics (2004) 57: 1627-1672.
|
Figures(8)
Antonio Tribuzio. Perturbations of minimizing movements and curves of maximal slope[J]. Networks and Heterogeneous Media, 2018, 13(3): 423-448. doi: 10.3934/nhm.2018019







 DownLoad:
DownLoad: