Citation: Nicola Gaetano Gatta, Rosaria Romano, Elenamaria Fioretti, Vittorio Gentile. Transglutaminase inhibition: possible therapeutic mechanisms to protect cells from death in neurological disorders[J]. AIMS Molecular Science, 2017, 4(4): 399-414. doi: 10.3934/molsci.2017.4.399

| [1] | Folk JE (1983) Mechanism and basis for specificity of transglutaminase-catalyzed e-(g-glutamyl) lysine bond formation. Adv Enzymol Relat Areas Mol Biol 54: 1-56. |
| [2] |
Lorand L, Conrad SM (1984) Transglutaminases. Mol Cell Biochem 58: 9-35. doi: 10.1007/BF00240602

|
| [3] | Piacentini M, Martinet N, Beninati S, et al. (1988) Free and protein conjugated-polyamines in mouse epidermal cells. Effect of high calcium and retinoic acid. J Biol Chem 263: 3790-3794. |
| [4] |
Song Y, Kirkpatrick LL, Schilling AB, et al. (2013) Transglutaminase and polyamination of tubulin: posttranslational modification for stabilizing axonal microtubules. Neuron 78: 109-123. doi: 10.1016/j.neuron.2013.01.036
|
| [5] | Achyuthan KE, Greenberg CS (1987) Identification of a guanosine triphosphate-binding site on guinea pig liver transglutaminase. Role of GTP and calcium ions in modulating activity. J Biol Chem 262: 1901-1906. |
| [6] |
Hasegawa G, Suwa M, Ichikawa Y, et al. (2003) A novel function of tissue-type transglutaminase: protein disulfide isomerase. Biochem J 373: 793-803. doi: 10.1042/bj20021084
|
| [7] | Lahav J, Karniel E, Bagoly Z, et al. (2009) Coagulation factor XIII serves as protein disulfide isomerase. Thromb Haemost 101: 840-844. |
| [8] |
Iismaa SE, Mearns BM, Lorand L, et al. (2009) Transglutaminases and disease: lessons from genetically engineered mouse models and inherited disorders. Physiol Rev 89: 991-1023. doi: 10.1152/physrev.00044.2008
|
| [9] |
Smethurst PA, Griffin M (1996) Measurement of tissue transglutaminase activity in a permeabilized cell system: its regulation by calcium and nucleotides. Biochem J 313: 803-808. doi: 10.1042/bj3130803
|
| [10] | Nakaoka H, Perez DM, Baek KJ, et al. (1994) Gh: a GTP-binding protein with transglutaminase activity and receptor signalling function. Science 264: 1593-1596. |
| [11] |
Gentile V, Porta R, Chiosi E, et al. (1997) Tissue transglutaminase and adenylate cyclase interactions in Balb-C 3T3 fibroblast membranes. Biochim Biophys Acta 1357: 115-122. doi: 10.1016/S0167-4889(97)00024-4
|
| [12] |
Nanda N, Iismaa SE, Owens WA, et al. (2001) Targeted inactivation of Gh/tissue transglutaminase II. J Biol Chem 276: 20673-20678. doi: 10.1074/jbc.M010846200
|
| [13] |
Mian S, El Alaoui S, Lawry J, et al. (1995) The importance of the GTP binding protein tissue transglutaminase in the regulation of cell cycle progression. FEBS Lett 370: 27-31. doi: 10.1016/0014-5793(95)00782-5
|
| [14] | Olaisen B, Gedde-Dahl TJR, Teisberg P, et al. (1985) A structural locus for coagulation factor XIIIA (F13A) is located distal to the HLA region on chromosome 6p in man. Am J Hum Genet 37: 215-220. |
| [15] | Yamanishi K, Inazawa J, Liew F-M, et al. (1992) Structure of the gene for human transglutaminase 1. J Biol Chem 267: 17858-17863. |
| [16] | Gentile V, Davies PJA, Baldini A (1994) The human tissue transglutaminase gene maps on chromosome 20q12 by in situ fluorescence hybridization. Genomics 20: 295-297. |
| [17] | Wang M, Kim IG, Steinert PM, et al. (1994) Assignment of the human transglutaminase 2 (TGM2) and transglutaminase 3 (TGM3) genes to chromosome 20q11.2. Genomics 23: 721-722. |
| [18] | Gentile V, Grant F, Porta R, et al. (1995) Human prostate transglutaminase is localized on chromosome 3p21.33-p22 by in situ fluorescence hybridization. Genomics 27: 219-220. |
| [19] | Grenard P, Bates MK, Aeschlimann D (2001) Evolution of transglutaminase genes: identification of a transglutaminases gene cluster on human chromosome 15q. Structure of the gene encoding transglutaminase X and a novel gene family member, transglutaminase Z. J Biol Chem 276: 33066-33078. |
| [20] |
Thomas H, Beck K, Adamczyk M, et al. (2013) Transglutaminase 6: a protein associated with central nervous system development and motor function. Amino Acids 44: 161-177. doi: 10.1007/s00726-011-1091-z
|
| [21] |
Bailey CDC, Johnson GVW (2004) Developmental regulation of tissue transglutaminase in the mouse forebrain. J Neurochem 91: 1369-1379. doi: 10.1111/j.1471-4159.2004.02825.x
|
| [22] | Kim SY, Grant P, Lee JHC, et al. (1999) Differential expression of multiple transglutaminases in human brain. Increased expression and cross-linking by transglutaminase 1 and 2 in Alzheimer's disease. J Biol Chem 274: 30715-30721. |
| [23] | lannaccone M, Giuberti G, De Vivo G, et al. (2013) Identification of a FXIIIA variant in human neuroblastoma cell lines. Int J Biochem Mol Biol 4: 102-107. |
| [24] |
Citron BA, Santa Cruz KS, Davies PJ, et al. (2001) Intron-exon swapping of transglutaminase mRNA and neuronal tau aggregation in Alzheimer's disease. J Biol Chem 276: 3295-3301. doi: 10.1074/jbc.M004776200
|
| [25] | De Laurenzi V, Melino G (2001) Gene disruption of tissue transglutaminase. Mol Cell Biol 21: 148-155. |
| [26] | Mastroberardino PG, Iannicola C, Nardacci R, et al. (2002) 'Tissue' transglutaminase ablation reduces neuronal death and prolongs survival in a mouse model of Huntington's disease. Cell Death Differ 9: 873-880. |
| [27] |
Lorand L, Graham RM (2003) Transglutaminases: crosslinking enzymes with pleiotropic functions. Nature Mol Cell Biol 4: 140-156. doi: 10.1038/nrm1014
|
| [28] |
Wolf J, Jäger C, Lachmann I, et al. (2013) Tissue transglutaminase is not a biochemical marker for Alzheimer's disease. Neurobiol Aging 34: 2495-2498. doi: 10.1016/j.neurobiolaging.2013.05.008
|
| [29] | Wilhelmus MMM, Drukarch B (2014) Tissue transglutaminase is a biochemical marker for Alzheimer's disease. Neurobiol Aging 35: 3-4. |
| [30] | Wolf J, Jäger C, Morawski M, et al. (2014) Tissue transglutaminase in Alzheimer's disease-facts and fiction: a reply to "Tissue transglutaminase is a biochemical marker for Alzheimer's disease". Neurobiol Aging 35: 5-9. |
| [31] | Adams RD, Victor M (1993) Principles of Neurology. |
| [32] |
Selkoe DJ, Abraham C, Ihara Y (1982) Alzheimer's disease: insolubility of partially purified paired helical filaments in sodium dodecyl sulfate and urea. Proc Natl Acad Sci USA 79: 6070-6074. doi: 10.1073/pnas.79.19.6070
|
| [33] |
Grierson AJ, Johnson GV, Miller CC (2001) Three different human isoforms and rat neurofilament light, middle and heavy chain proteins are cellular substrates for transglutaminase. Neurosci Lett 298: 9-12. doi: 10.1016/S0304-3940(00)01714-6
|
| [34] |
Singer SM, Zainelli GM, Norlund MA (2002) Transglutaminase bonds in neurofibrillary tangles and paired helical filament t early in Alzheimer's disease. Neurochem Int 40: 17-30. doi: 10.1016/S0197-0186(01)00061-4
|
| [35] |
Halverson RA, Lewis J, Frausto S, et al. (2005) Tau protein is cross-linked by transglutaminase in P301L tau transgenic mice. J Neurosci 25: 1226-33. doi: 10.1523/JNEUROSCI.3263-04.2005
|
| [36] |
Jeitner TM, Matson WR, Folk JE, et al. (2008) Increased levels of g-glutamylamines in Huntington disease CSF. J Neurochem 106: 37-44. doi: 10.1111/j.1471-4159.2008.05350.x
|
| [37] |
Dudek SM, Johnson GV (1994) Transglutaminase facilitates the formation of polymers of the beta-amyloid peptide. Brain Res 651: 129-33. doi: 10.1016/0006-8993(94)90688-2
|
| [38] |
Hartley DM, Zhao C, Speier AC, et al. (2008) Transglutaminase induces protofibril-like amyloid b protein assemblies that are protease-resistant and inhibit long-term potentiation. J Biol Chem 283: 16790-16800. doi: 10.1074/jbc.M802215200
|
| [39] |
Citron BA, Suo Z, SantaCruz K, et al. (2002) Protein crosslinking, tissue transglutaminase, alternative splicing and neurodegeneration. Neurochem Int 40: 69-78. doi: 10.1016/S0197-0186(01)00062-6
|
| [40] |
Junn E, Ronchetti RD, Quezado MM, et al. (2003) Tissue transglutaminase-induced aggregation of a-synuclein: Implications for Lewy body formation in Parkinson's disease and dementia with Lewy bodies. Proc Natl Acad Sci USA 100: 2047-2052. doi: 10.1073/pnas.0438021100
|
| [41] |
Zemaitaitis MO, Lee JM, Troncoso JC, et al (2000) Transglutaminase-induced cross-linking of tau proteins in progressive supranuclear palsy. J Neuropathol Exp Neurol 59: 983-989. doi: 10.1093/jnen/59.11.983
|
| [42] |
Zemaitaitis MO, Kim SY, Halverson RA, et al. (2003) Transglutaminase activity, protein, and mRNA expression are increased in progressive supranuclear palsy. J Neuropathol Exp Neurol 62: 173-184. doi: 10.1093/jnen/62.2.173
|
| [43] |
Iuchi S, Hoffner G, Verbeke P, et al. (2003) Oligomeric and polymeric aggregates formed by proteins containing expanded polyglutamine. Proc Natl Acad Sci USA 100: 2409-2414. doi: 10.1073/pnas.0437660100
|
| [44] |
Gentile V, Sepe C, Calvani M, et al. (1998) Tissue transglutaminase-catalyzed formation of high-molecular-weight aggregates in vitro is favored with long polyglutamine domains: a possible mechanism contributing to CAG-triplet diseases. Arch Biochem Biophys 352: 314-321. doi: 10.1006/abbi.1998.0592
|
| [45] |
Kahlem P, Green H, Djian P (1998) Transglutaminase action imitates Huntington's disease: selective polymerization of huntingtin containing expanded polyglutamine. Mol Cell 1: 595-601. doi: 10.1016/S1097-2765(00)80059-3
|
| [46] |
Karpuj MV, Garren H, Slunt H, et al (1999) Transglutaminase aggregates huntingtin into nonamyloidogenic polymers, and its enzymatic activity increases in Huntington's disease brain nuclei. Proc Natl Acad Sci USA 96: 7388-7393. doi: 10.1073/pnas.96.13.7388
|
| [47] |
Segers-Nolten IM, Wilhelmus MM, Veldhuis G, et al. (2008) Tissue transglutaminase modulates α-synuclein oligomerization. Protein Sci 17: 1395-1402. doi: 10.1110/ps.036103.108
|
| [48] |
Lai TS, Tucker T, Burke JR, et al. (2004) Effect of tissue transglutaminase on the solubility of proteins containing expanded polyglutamine repeats. J Neurochem 88: 1253-1260. doi: 10.1046/j.1471-4159.2003.02249.x
|
| [49] |
Konno T, Mori T, Shimizu H, et al. (2005) Paradoxical inhibition of protein aggregation and precipitation by transglutaminase-catalyzed intermolecular cross-linking. J Biol Chem 280: 17520-17525. doi: 10.1074/jbc.M413988200
|
| [50] |
The Huntington's Disease Collaborative Research Group (1993) A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington's disease chromosome. Cell 72: 971-983. doi: 10.1016/0092-8674(93)90585-E
|
| [51] |
Banfi S, Chung MY, Kwiatkowski TJ, et al. (1993) Mapping and cloning of the critical region for the spinocerebellar ataxia type 1 gene (SCA1) in a yeast artificial chromosome contig spanning 1.2 Mb. Genomics 18: 627-635. doi: 10.1016/S0888-7543(05)80365-9
|
| [52] |
Sanpei K, Takano H, Igarashi S, et al. (1996) Identification of the spinocerebellar ataxia type 2 gene using a direct identification of repeat expansion and cloning technique, DIRECT. Nat Genet 14: 277-284. doi: 10.1038/ng1196-277
|
| [53] | Pujana MA, Volpini V, Estivill X (1998) Large CAG/CTG repeat templates produced by PCR, usefulness for the DIRECT method of cloning genes with CAG/CTG repeat expansions. Nucleic Acids Res 1: 1352-1353. |
| [54] |
Fletcher CF, Lutz CM, O'Sullivan TN, et al. (1996) Absence epilepsy in tottering mutant mice is associated with calcium channel defects. Cell 87: 607-617. doi: 10.1016/S0092-8674(00)81381-1
|
| [55] |
Vincent JB, Neves-Pereira ML, Paterson AD, et al. (2000) An unstable trinucleotide-repeat region on chromosome 13 implicated in spinocerebellar ataxia: a common expansion locus. Am J Hum Genet 66: 819-829. doi: 10.1086/302803
|
| [56] |
Holmes SE, O'Hearn E, Margolis RL (2003) Why is SCA12 different from other SCAs? Cytogenet Genome Res 100: 189-197. doi: 10.1159/000072854
|
| [57] |
Imbert G, Trottier Y, Beckmann J, et al. (1994) The gene for the TATA binding protein (TBP) that contains a highly polymorphic protein coding CAG repeat maps to 6q27. Genomics 21: 667-668. doi: 10.1006/geno.1994.1335
|
| [58] |
La Spada AR, Wilson EM, Lubahn DB, et al. (1991) Androgen receptor gene mutations in X-linked spinal and bulbar muscular atrophy. Nature 352: 77-79. doi: 10.1038/352077a0
|
| [59] | Onodera O, Oyake M, Takano H, et al. (1995) Molecular cloning of a full-length cDNA for dentatorubral-pallidoluysian atrophy and regional expressions of the expanded alleles in the CNS. Am J Hum Genet 57: 1050-1060. |
| [60] | Cooper AJL, Sheu K-FR, Burke JR, et al. (1999) Pathogenesis of inclusion bodies in (CAG)n/Qn-expansion diseases with special reference to the role of tissue transglutaminase and to selective vulnerability. J Neurochem 72: 889-899. |
| [61] |
Hadjivassiliou M, Maki M, Sanders DS, et al. (2006) Autoantibody targeting of brain and intestinal transglutaminase in gluten ataxia. Neurology 66: 373-377. doi: 10.1212/01.wnl.0000196480.55601.3a
|
| [62] |
Boscolo S, Lorenzon A, Sblattero D, et al. (2010) Anti transglutaminase antibodies cause ataxia in mice. Plos One 5: e9698. doi: 10.1371/journal.pone.0009698
|
| [63] |
Stamnaes J, Dorum S, Fleckenstein B, et al. (2010) Gluten T cell epitope targeting by TG3 and TG6; implications for dermatitis herpetiformis and gluten ataxia. Amino Acids 39: 1183-1191. doi: 10.1007/s00726-010-0554-y
|
| [64] | Lerner A, Matthias T (2016) GUT-the Trojan horse in remote organs' autoimmunity. J Clin Cell Immunol 7: 401. |
| [65] |
Matthias T, Jeremias P, Neidhofer S, et al. (2016) The industrial food additive microbial transglutaminase, mimics the tissue transglutaminase and is immunogenic in celiac disease patients. Autoimmun Rev 15: 1111-1119. doi: 10.1016/j.autrev.2016.09.011
|
| [66] | Lerner A, Neidhofer S, Matthias T (2015) Transglutaminase 2 and anti transglutaminase 2 autoantibodies in celiac disease and beyond: Part A: TG2 double-edged sword: gut and extraintestinal involvement. Immunome Res 11: 101-105. |
| [67] |
Wakshlag JJ, Antonyak MA, Boehm JE, et al. (2006) Effects of tissue transglutaminase on beta-amyloid 1-42-induced apoptosis. Protein J 25: 83-94. doi: 10.1007/s10930-006-0009-1
|
| [68] |
Lee JH, Jeong J, Jeong EM, et al. (2014) Endoplasmic reticulum stress activates transglutaminase 2 leading to protein aggregation. Int J Mol Med 33: 849-855. doi: 10.3892/ijmm.2014.1640
|
| [69] |
Grosso H, Woo JM, Lee KW, et al. (2014) Transglutaminase 2 exacerbates α-synuclein toxicity in mice and yeast. FASEB J 28: 4280-4291. doi: 10.1096/fj.14-251413
|
| [70] |
Zhang J, Wang S, Huang W, et al. (2016) Tissue transglutaminase and its product isopeptide are increased in Alzheimer's disease and APPswe/PS1dE9 double transgenic mice brains. Mol Neurobiol 53: 5066-5078. doi: 10.1007/s12035-015-9413-x
|
| [71] |
Wilhelmus MM, De JM, Smit AB, et al (2016) Catalytically active tissue transglutaminase colocalises with Ab pathology in Alzheimer's disease mouse models. Sci Rep 6: 20569. doi: 10.1038/srep20569
|
| [72] |
Wilhelmus MMM, De JM, Rozemuller AJM, et al. (2012) Transglutaminase 1 and its regulator Tazarotene-induced gene 3 localize to neuronal tau inclusions in tauopathies. J Pathol 226: 132-142. doi: 10.1002/path.2984
|
| [73] | Basso M, Berlin J, Xia L, et al. (2012) Transglutaminase inhibition protects against oxidative stress-induced neuronal death downstream of pathological ERK activation. J Neurosci 39: 6561-6569. |
| [74] |
Lee J, Kim YS, Choi DH, et al. (2004) Transglutaminase 2 induces nuclear factor-kB activation via a novel pathway in BV-2 microglia. J Biol Chem 279: 53725-53735. doi: 10.1074/jbc.M407627200
|
| [75] | Kumar S, Mehta K (2012) Tissue transglutaminase constitutively activates HIF-1a promoter and nuclear factor-kB via a non-canonical pathway. Plos One 7: e49321 |
| [76] |
Lu S, Saydak M, Gentile V, et al. (1995) Isolation and characterization of the human tissue transglutaminase promoter. J Biol Chem 270: 9748-9755. doi: 10.1074/jbc.270.17.9748
|
| [77] |
Ientile R, Currò M, Caccamo D (2015) Transglutaminase 2 and neuroinflammation. Amino Acids 47: 19-26. doi: 10.1007/s00726-014-1864-2
|
| [78] |
Griffith OW, Larsson A, Meister A (1977) Inhibition of g-glutamylcysteine synthetase by cystamine: an approach to a therapy of 5-oxoprolinuria (pyroglutamic aciduria). Biochem Biophys Res Commun 79: 919-925. doi: 10.1016/0006-291X(77)91198-6
|
| [79] |
Igarashi S, Koide R, Shimohata T, et al. (1998) Suppression of aggregate formation and apoptosis by transglutaminase inhibitors in cells expressing truncated DRPLA protein with an expanded polyglutamine stretch. Nat Genet 18: 111-117. doi: 10.1038/ng0298-111
|
| [80] | Karpuj MV, Becher MW, Springer JE, et al. (2002) Prolonged survival and decreased abnormal movements in transgenic model of Huntington disease, with administration of the transglutaminase inhibitor cystamine. Nat Med 8: 143-149. |
| [81] | Dedeoglu A, Kubilus JK, Jeitner TM, et al. (2002) Therapeutic effects of cystamine in a murine model of Huntington's disease. J Neurosci 22: 8942-8950. |
| [82] | Lesort M, Lee M, Tucholski J, et al. (2003) Cystamine inhibits caspase activity. Implications for the treatment of polyglutamine disorders. J Biol Chem 278: 3825-3830. |
| [83] |
Dubinsky R, Gray C (2006) CYTE-I-HD: Phase I Dose Finding and Tolerability Study of Cysteamine (Cystagon) in Huntington's Disease. Movement Disord 21: 530-533. doi: 10.1002/mds.20756
|
| [84] |
Langman CB, Greenbaum LA, Sarwal M, et al. (2012) A randomized controlled crossover trial with delayed-release cysteamine bitartrate in nephropathic cystinosis: effectiveness on white blood cell cystine levels and comparison of safety. Clin J Am Soc Nephrol 7: 1112-1120. doi: 10.2215/CJN.12321211
|
| [85] |
Besouw M, Masereeuw R, Van DHL, et al. (2013) Cysteamine: an old drug with new potential. Drug Discov Today 18: 785-792. doi: 10.1016/j.drudis.2013.02.003
|
| [86] |
Hadjivassiliou M, Aeschlimann P, Strigun A, et al. (2008) Autoantibodies in gluten ataxia recognize a novel neuronal transglutaminase. Ann Neurol 64: 332-343. doi: 10.1002/ana.21450
|
| [87] |
Krasnikov BF, Kim SY, McConoughey SJ, et al. (2005) Transglutaminase activity is present in highly purified nonsynaptosomal mouse brain and liver mitochondria. Biochemistry 44: 7830-7843. doi: 10.1021/bi0500877
|
| [88] |
Menalled LB, Kudwa AE, Oakeshott S, et al. (2014) Genetic deletion of transglutaminase 2 does not rescue the phenotypic deficits observed in R6/2 and zQ175 mouse models of Huntington's disease. Plos One 9: e99520-e99520. doi: 10.1371/journal.pone.0099520
|
| [89] |
Bailey CD, Johnson GV (2005) Tissue transglutaminase contributes to disease progression in the R6/2 Huntington's disease mouse model via aggregate-independent mechanisms. J Neurochem 92: 83-92. doi: 10.1111/j.1471-4159.2004.02839.x
|
| [90] | Davies JE, Rose C, Sarkar S, et al. (2010) Cystamine suppresses polyalanine toxicity in a mouse model of oculopharyngeal muscular dystrophy. Sci Transl Med 2: 34-40. |
| [91] |
Keillor JW, Apperley KYP (2016) Transglutaminase inhibitors: a patent review. Expert Opin Ther Pat 26: 49-63. doi: 10.1517/13543776.2016.1115836
|
| [92] |
Song M, Hwang H, Im CY, et al. (2017) Recent progress in the development of Transglutaminase 2 (TGase2) inhibitors. J Med Chem 60: 554-567. doi: 10.1021/acs.jmedchem.6b01036
|
| [93] |
Pietsch M, Wodtke R, Pietzsch J, et al. (2013) Tissue transglutaminase: An emerging target for therapy and imaging. Bioorg Med Chem Lett 23: 6528-6543. doi: 10.1016/j.bmcl.2013.09.060
|
| [94] |
Bhatt MP, Lim YC, Hwang J, et al. (2013) C-peptide prevents hyperglycemia-induced endothelial apoptosis through inhibition of reactive oxygen species-mediated transglutaminase 2 activation. Diabetes 62: 243-253. doi: 10.2337/db12-0293
|
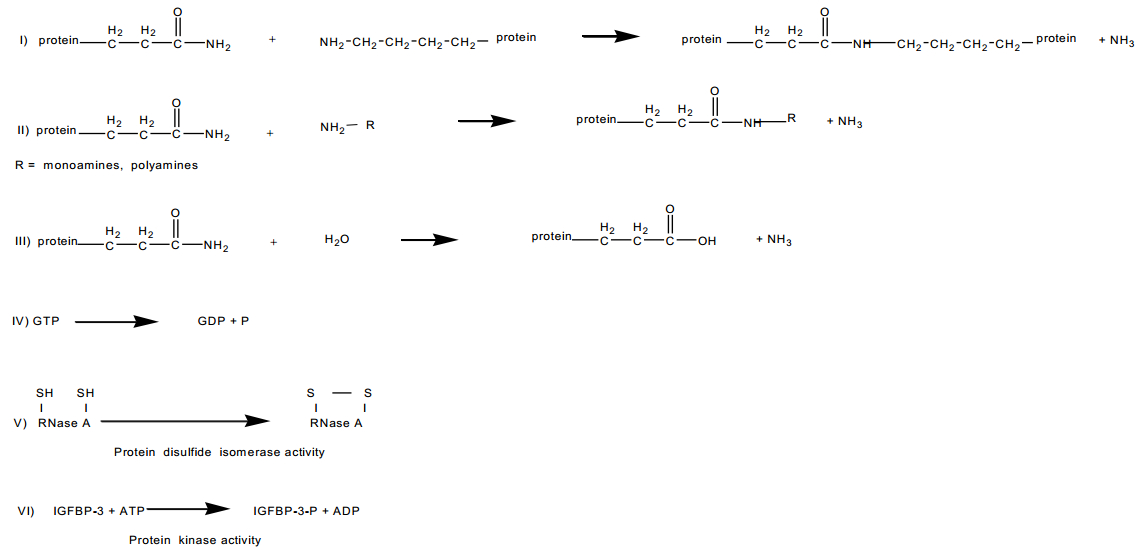
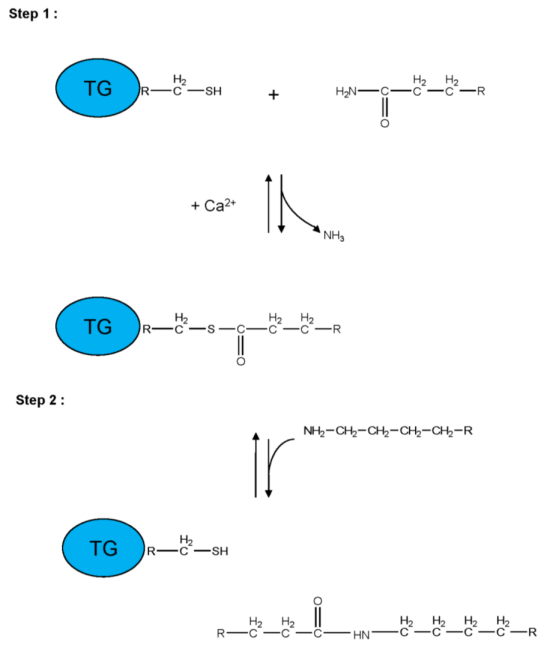
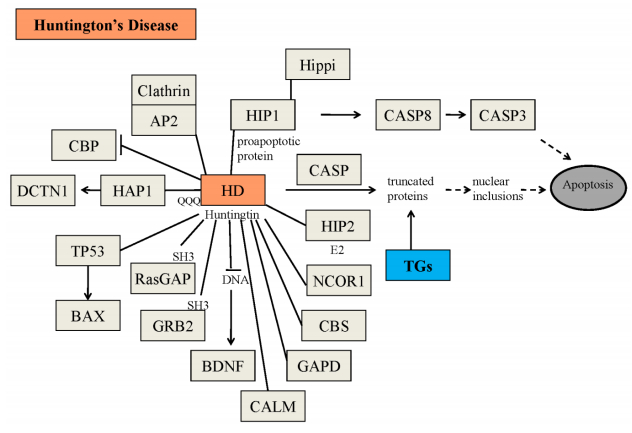
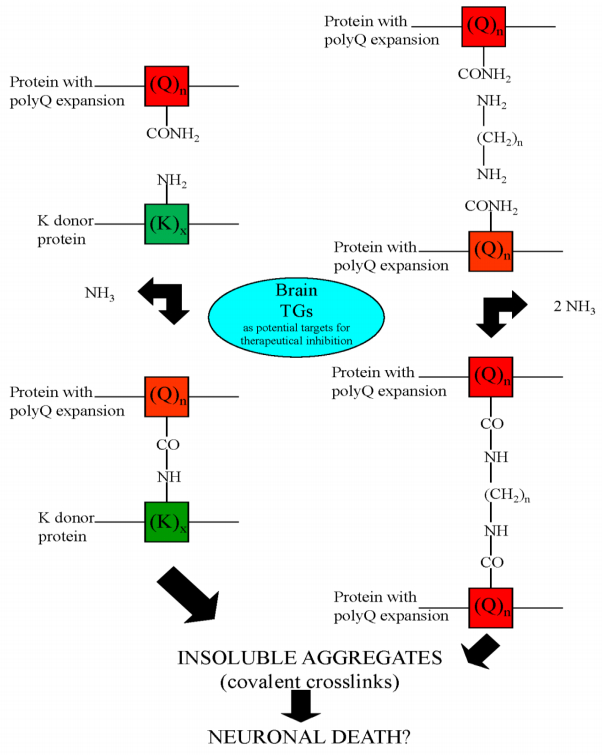
Figures(5) / Tables(3)

Nicola Gaetano Gatta, Rosaria Romano, Elenamaria Fioretti, Vittorio Gentile. Transglutaminase inhibition: possible therapeutic mechanisms to protect cells from death in neurological disorders[J]. AIMS Molecular Science, 2017, 4(4): 399-414. doi: 10.3934/molsci.2017.4.399







 DownLoad:
DownLoad: