Tuberculosis is the leading cause of death worldwide from a single infectious agent; it has also been declared a threat to humanity by the World Health Organization. New insights indicate that the innate immune response plays a crucial role in determining the outcome of the infection. In this study, we assessed the role of macrophages in the innate immune response through a simple mathematical model. Our results confirm that macrophages provide the primary protective response against Mycobacterium tuberculosis. However, they also highlight the importance of other innate cells in the outcome of infection. Specifically, our findings suggest that, in addition to macrophage activity, the involvement of other innate immune cells is essential for eliminating or controlling bacterial progression, ultimately leading to an adaptive immune response.
Citation: Eduardo Ibargüen-Mondragón, M. Victoria Otero-Espinar, Miller Cerón Gómez. A within-host model on the interaction dynamics between innate immune cells and Mycobacterium tuberculosis[J]. Mathematical Biosciences and Engineering, 2025, 22(3): 511-527. doi: 10.3934/mbe.2025019
Tuberculosis is the leading cause of death worldwide from a single infectious agent; it has also been declared a threat to humanity by the World Health Organization. New insights indicate that the innate immune response plays a crucial role in determining the outcome of the infection. In this study, we assessed the role of macrophages in the innate immune response through a simple mathematical model. Our results confirm that macrophages provide the primary protective response against Mycobacterium tuberculosis. However, they also highlight the importance of other innate cells in the outcome of infection. Specifically, our findings suggest that, in addition to macrophage activity, the involvement of other innate immune cells is essential for eliminating or controlling bacterial progression, ultimately leading to an adaptive immune response.

| [1] | Global tuberculosis report 2023, World Health Organization, 2023. Available from: https://www.who.int/publications/i/item/9789240083851 |
| [2] |
D. D. Chaplin, Overview of the immune response, J. Allergy Clin. Immunol., 125 (2010), S3–S23. https://doi.org/10.1016/j.jaci.2009.12.980 doi: 10.1016/j.jaci.2009.12.980

|
| [3] |
P. Sankar, B. B. Mishra, Early innate cell interactions with Mycobacterium tuberculosis in protection and pathology of tuberculosis, Front. Immunol., 14 (2023), 1–21. https://doi.org/10.3389/fimmu.2023.1260859 doi: 10.3389/fimmu.2023.1260859
|
| [4] | M. M. Ravesloot-Chávez, E. Van Dis, S. A. Stanley, The innate immune responseto mycobacterium tuberculosis infection, Annu. Rev. Immunol., 39 (2021), 611–37. https://10.1146/annurev-immunol-093019-010426 |
| [5] |
S. H. E. Kaufmann, How can immunology contribuite to the control of tuberculosis?, Nat. Rev. Immunol., 1 (2001), 20–30. https://doi.org/10.1038/35095558 doi: 10.1038/35095558
|
| [6] |
T. R. Lerner, S. Borel, M. G. Gutierrez, The innate immune response in human tuberculosis, Cell. Microbio., 17 (2015), 1277–1285. https://doi.org/10.1111/cmi.12480 doi: 10.1111/cmi.12480
|
| [7] |
E. Guirado, L. S. Schlesinger, G. Kaplan, Macrophages in tuberculosis: Friend or foe, Semin. Immunol., 35 (2013), 563–583. https://doi.org/10.1007/s00281-013-0388-2 doi: 10.1007/s00281-013-0388-2
|
| [8] |
A. Vu, I. Glassman, G. Campbell, S. Yeganyan, J. Nguyen, A. Shin, et al., Host cell death andmodulation of immune response against mycobacterium tuberculosis infection, Int. J. Mol. Sci., 25 (2021), 1–22. https://doi.org/10.3390/ijms25116255 doi: 10.3390/ijms25116255
|
| [9] |
J. K. Kim, P. Silwal, E. K. Jo, Host-Pathogen dialogues in autophagy, apoptosis, and necrosis during Mycobacterial infection, Immune Netw., 20 (2020), 1–15. https://doi.org/10.4110/in.2020.20.e37 doi: 10.4110/in.2020.20.e37
|
| [10] |
D. M. Kelly, A. M. Ten Bokum, S. M. O'Leary, M. P. O'Sullivan, J. Keane, Bystander macrophage apoptosis after Mycobacterium tuberculosis H37Ra infection, Infect. Immun., 76 (2008), 351–360. https://doi.org/10.1128/IAI.00614-07 doi: 10.1128/IAI.00614-07
|
| [11] |
F. J. Roca, L. J. Whitworth, S. Redmond, A. A. Jones, L. Ramakrishnan, J. Keane, TNF induces pathogenic programmed macrophage necrosis in tuberculosis through a mitochondrial-lysosomal-endoplasmic reticulum circuit, Cell, 178 (2019), 1344–1361. https://doi.org/10.1016/j.cell.2019.08.004 doi: 10.1016/j.cell.2019.08.004
|
| [12] |
F. Ahmad, A. Rani, A. Alam, S. Zarin, S. Pandey, H. Singh, et al., Macrophage: A cell with many faces and functions in tuberculosis, Front. Immunol., 13 (2022), 1–18. https://doi.org/10.3389/fimmu.2022.747799 doi: 10.3389/fimmu.2022.747799
|
| [13] |
J. Pieters, Mycobacterium tuberculosis and the macrophage: Maintaining a balance, Cell Host Microbe, 3 (2008), 399–407. https://doi.org/10.1016/j.chom.2008.05.006 doi: 10.1016/j.chom.2008.05.006
|
| [14] |
J. A. Philips, J. P. Ernst, Tuberculosis pathogenesis and immunity, Annu. Rev. Pathol. Mech. Dis., 7 (2012), 353–384. https://doi.org/10.1146/annurev-pathol-011811-132458 doi: 10.1146/annurev-pathol-011811-132458
|
| [15] |
H. Kim, S. J. Shin, Pathological and protective roles of dendritic cells in Mycobacterium tuberculosis infection: Interaction between host immune responses and pathogen evasion, Front. Cell. Infect. Microbiol., 12 (2022), 1–27. https://doi.org/10.3389/fcimb.2022.891878 doi: 10.3389/fcimb.2022.891878
|
| [16] |
D. Kirschner, E. Pienaar, S. Marino, J. J. Linderman, A review of computational and mathematical modeling contributions to our understanding of Mycobacterium tuberculosis within-host infection and treatment, Curr. Opin. Syst. Biol., 3 (2017), 170–185. https://doi.org/10.1016/j.coisb.2017.05.014 doi: 10.1016/j.coisb.2017.05.014
|
| [17] |
D. Chakraborty, S. Batabyal, V. V. Ganusov, A brief overview of mathematical modeling of the within-host dynamics of Mycobacterium tuberculosis, Front. Appl. Math. Stat., 10 (2024), 1355–373. https://doi.org/10.3389/fams.2024.1355373 doi: 10.3389/fams.2024.1355373
|
| [18] |
E. Ibargüen-Mondragón, L. Esteva, L. Chávez-Galán, A mathematical model for cellular immunology of tuberculosis, Math. Biosci. Eng., 11 (2011), 973–986. https://doi.org/10.3934/mbe.2011.8.973 doi: 10.3934/mbe.2011.8.973
|
| [19] |
E. Ibargüen-Mondragón, L. Esteva, E. M. Burbano-Rosero, Mathematical model for the growth of Mycobacterium tuberculosis in the granuloma, Math. Biosci. Eng., 15 (2018), 407–428. https://doi.org/10.3934/mbe.2018018 doi: 10.3934/mbe.2018018
|
| [20] | E. Ibarguen-Mondragon, L. Esteva, L. Chávez-Galán, Estabilidad global para un modelo matemático sobre la respuesta inmune innata de macrófagos contra el Micobacterium tuberculosis, Rev. Sigma., 10 (2010), 1–17. |
| [21] |
G. Pedruzzi, K. V. Rao, S. Chatterjee, Mathematical model of mycobacterium–host interaction describes physiology of persistence, J. Theor. Biol., 376 (2015), 105–117. https://doi.org/10.1016/j.jtbi.2015.03.031 doi: 10.1016/j.jtbi.2015.03.031
|
| [22] |
D. Gammack, C. R. Doering, D. E. Kirschner, Macrophage response to Mycobacterium tuberculosis infection, J. Math. Biol., 48 (2004), 218–242. https://doi.org/10.1007/s00285-003-0232-8 doi: 10.1007/s00285-003-0232-8
|
| [23] |
F. Clarelli, R. Natalini, A pressure model of immune response to mycobacteriumtuberculosis infection in several space dimensions, Math. Biosci. Eng., 7 (2004), 277–300. https://doi.org/10.3934/mbe.2010.7.277 doi: 10.3934/mbe.2010.7.277
|
| [24] |
E. Pienaar, M. Lerm, A mathematical model of the initial interaction between Mycobacterium tuberculosis and macrophages, J. Theor. Biol., 342 (2014), 23–32. http://dx.doi.org/10.1016/j.jtbi.2013.09.029 doi: 10.1016/j.jtbi.2013.09.029
|
| [25] |
R. V. Carvalho, J. Kleijn, A. H. Meijer, F. J. Verbeek, Modelinginnate immune response to early mycobacterium infection, Comput. Math. Methods Med., 2012 (2012), 1–12. https://doi.org/10.1155/2012/790482 doi: 10.1155/2012/790482
|
| [26] |
L. R. Joslyn, J. J. Linderman, D. E. Kirschner, A virtual host model of Mycobacterium tuberculosis infection identifies early immune events as predictive of infection outcomes, J. Theor. Biol., 539 (2022), 1–15. https://doi.org/10.1016/j.jtbi.2022.111042 doi: 10.1016/j.jtbi.2022.111042
|
| [27] |
A. Nisa, F. C. Kipper, D. Panigrahy, S. Tiwari, A. Kupz, S. Subbian, Different modalities of host cell death and their impact on Mycobacterium tuberculosis infection, Am. J. Physiol. Cell Physiol., 323 (2022), 1444–1474. https://doi.org/10.1152/ajpcell.00246.2022 doi: 10.1152/ajpcell.00246.2022
|
| [28] |
M. Divangahi, S. M. Behar, H. Remold, Dying to live: How the death modality of the infected macrophage affects immunity to tuberculosis, Adv. Exp. Med. Biol., 783 (2013), 103–120. https://doi.org/10.1007/978-1-4614-6111-1_6 doi: 10.1007/978-1-4614-6111-1_6
|
| [29] |
D. Mahamed, M. Boulle, Y. Ganga, C. M. Arthur, S. Skroch, L. Oom, et al., Intracellular growth of Mycobacterium tuberculosis after macrophage cell death leads to serial killing of host cells, Elife, 6 (2017), 1–26. https://doi.org/10.7554/eLife.22028 doi: 10.7554/eLife.22028
|
| [30] | L. Perko, Differential Equations and Dynamical Systems, 2$^{nd}$ edition, Springer Science & Business Media, New York, 2013. https://doi.org/10.1007/978-1-4613-0003-8 |
| [31] |
D. Sud, C. Bigbee, J. L. Flynn, D. E. Kirschner, Contribution of CD8+ T cells to control of Mycobacterium tuberculosis infection, J. Immun., 176 (2006), 4296–4314. https://doi.org/10.4049/jimmunol.176.7.4296 doi: 10.4049/jimmunol.176.7.4296
|
| [32] |
F. Krombach, S. Münzing, A. M. Allmeling, J. T. Gerlach, J. Behr, M. Dörger, Cell size of alveolar macrophages: An interspecies comparison, J. Immun., 105 (1997), 1261–1263. https://doi.org/10.1289/ehp.97105s51261 doi: 10.1289/ehp.97105s51261
|
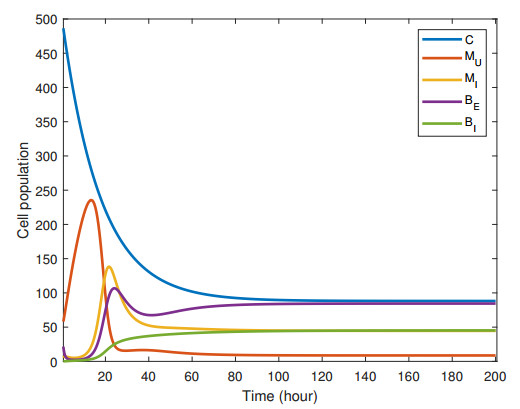
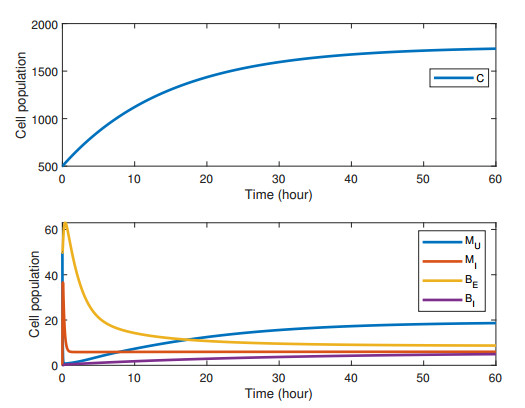
Figures(2) / Tables(4)

Eduardo Ibargüen-Mondragón, M. Victoria Otero-Espinar, Miller Cerón Gómez. A within-host model on the interaction dynamics between innate immune cells and Mycobacterium tuberculosis[J]. Mathematical Biosciences and Engineering, 2025, 22(3): 511-527. doi: 10.3934/mbe.2025019







 DownLoad:
DownLoad: