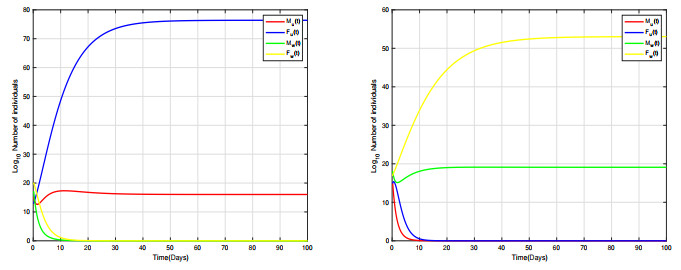
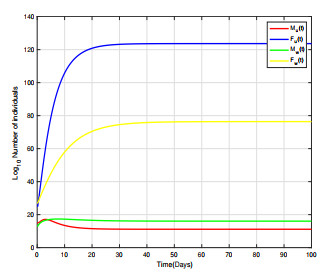
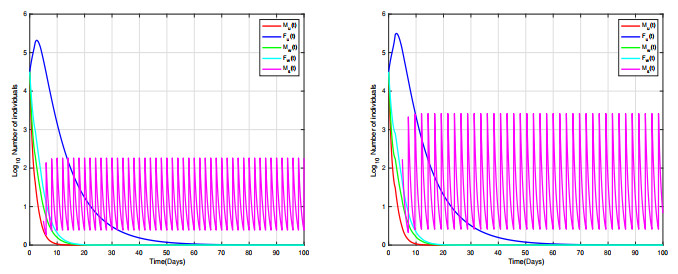
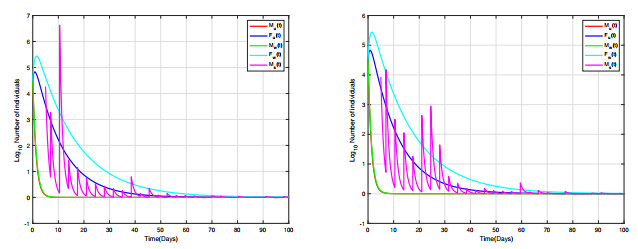
Combining Sterile and Incompatible Insect techniques can significantly reduce mosquito populations and prevent the transmission of diseases between insects and humans. This paper describes impulsive differential equations for the control of a mosquito with Wolbachia. Several interesting conditions are created when sterile male mosquitoes are released impulsively, ensuring both open- and closed-loop control. To determine the wild mosquito population size in real-time, we propose an open-loop control system, which uses impulsive and constant releases of sterile male mosquitoes. A closed-loop control scheme is also being investigated, which specifies the release of sterile mosquitoes according to the size of the wild mosquito population. To eliminate or reduce a mosquito population below a certain threshold, the Sterile insect technique involves mass releases of sterile insects. Numerical simulations verify the theoretical results.
Citation: Rajivganthi Chinnathambi, Fathalla A. Rihan. Analysis and control of Aedes Aegypti mosquitoes using sterile-insect techniques with Wolbachia[J]. Mathematical Biosciences and Engineering, 2022, 19(11): 11154-11171. doi: 10.3934/mbe.2022520
Combining Sterile and Incompatible Insect techniques can significantly reduce mosquito populations and prevent the transmission of diseases between insects and humans. This paper describes impulsive differential equations for the control of a mosquito with Wolbachia. Several interesting conditions are created when sterile male mosquitoes are released impulsively, ensuring both open- and closed-loop control. To determine the wild mosquito population size in real-time, we propose an open-loop control system, which uses impulsive and constant releases of sterile male mosquitoes. A closed-loop control scheme is also being investigated, which specifies the release of sterile mosquitoes according to the size of the wild mosquito population. To eliminate or reduce a mosquito population below a certain threshold, the Sterile insect technique involves mass releases of sterile insects. Numerical simulations verify the theoretical results.

| [1] |
H. Hughes, N. F. Britton, Modelling the use of Wolbachia to control dengue fever transmission, Bull. Math. Biol., 75 (2013), 796–818. https://doi.org/10.1007/s11538-013-9835-4 doi: 10.1007/s11538-013-9835-4

|
| [2] |
Y. Hui, J. Yu, Global asymptotic stability in a non-autonomous delay mosquito population suppression model, Appl. Math. Lett., 124 (2022), 107599. https://doi.org/10.1016/j.aml.2021.107599 doi: 10.1016/j.aml.2021.107599
|
| [3] |
M. Z. Ndii, R. I. Hickson, D. Allingham, G. N. Mercer, Modelling the transmission dynamics of dengue in the presence of Wolbachia, Math. Biosci., 262 (2015), 157–166. https://doi.org/10.1016/j.mbs.2014.12.011 doi: 10.1016/j.mbs.2014.12.011
|
| [4] |
P. A. Bliman, M. S. Aronna, F. C. Coelho, M. A. H. B. Silva, Ensuring successful introduction of Wolbachia in natural populations of Aedes Aegypti by means of feedback control, J. Math. Biol., 76 (2018), 1269–1300. https://doi.org/10.1007/s00285-017-1174-x doi: 10.1007/s00285-017-1174-x
|
| [5] |
L. Multerer, T. Smith, N. Chitnis, Modeling the impact of sterile males on an Aedes Aegypti population with optimal control, Math. Biosci., 311 (2019), 91–102. https://doi.org/10.1016/j.mbs.2019.03.003 doi: 10.1016/j.mbs.2019.03.003
|
| [6] |
B. Zheng, L. Chen, Q. Sun, Analyzing the control of dengue by releasing Wolbachia infected male mosquitoes through a delay differential equation model, Math. Biosci. Eng., 16 (2019), 5531–5550. http://dx.doi.org/10.3934/mbe.2019275 doi: 10.3934/mbe.2019275
|
| [7] | F. A. Rihan, Delay Differential Equations and Applications to Biology, Springer, Singapore, 2021. https://doi.org/10.1007/978-981-16-0626-7 |
| [8] |
A. Aghriche, R. Yafia, M. A. A. Alaoui, A. Tridane, F. A. Rihan, Oscillations induced by quiescent adult female in a reaction-diffusion model of wild Aedes Aegypti mosquitoes, Int. J. Bifurcation Chaos, 29 (2019), 1950189. https://doi.org/10.1142/S021812741950189X doi: 10.1142/S021812741950189X
|
| [9] |
S. P. Sinkins, Wolbachia and cytoplasmic incompatibility in mosquitoes, Insect Biochem. Mol. Biol., 34 (2004), 723–729. https://doi.org/10.1016/j.ibmb.2004.03.025 doi: 10.1016/j.ibmb.2004.03.025
|
| [10] |
I. Iturbe-Ormaetxe, T. Walker, S. L. O'Neill, Wolbachia and the biological control of mosquito-borne disease, EMBO Rep., 12 (2011), 508–518. https://doi.org/10.1038/embor.2011.84 doi: 10.1038/embor.2011.84
|
| [11] |
X. Zhang, S. Tang, R. A. Cheke, H. Zhu, Modeling the effects of augmentation strategies on the control of dengue fever With an impulsive differential equation, Bull. Math. Biol., 78 (2016), 1968–2010. https://doi.org/10.1007/s11538-016-0208-7 doi: 10.1007/s11538-016-0208-7
|
| [12] |
Y. Li, X. Liu, A sex-structured model with birth pulse and release strategy for the spread of Wolbachia in mosquito population, J. Theor. Biol., 448 (2018), 53–65. https://doi.org/10.1016/j.jtbi.2018.04.001 doi: 10.1016/j.jtbi.2018.04.001
|
| [13] |
B. Zheng, M. Tang, J. Yu, Modeling Wolbachia spread in mosquitoes through delay differential equations, SIAM J. Appl. Math., 74 (2014), 743–770. https://doi.org/10.1137/13093354X doi: 10.1137/13093354X
|
| [14] |
D. Li, H. Wan, The threshold infection level for Wolbachia invasion in a two sex mosquito population model, Bull. Math. Biol., 81 (2019), 2596–2624. https://doi.org/10.1007/s11538-019-00620-1 doi: 10.1007/s11538-019-00620-1
|
| [15] |
Z. Zhang, B. Zheng, Dynamics of a mosquito population suppression model with a saturated Wolbachia release rate, Appl. Math. Lett., 129 (2022), 107933. https://doi.org/10.1016/j.aml.2022.107933 doi: 10.1016/j.aml.2022.107933
|
| [16] |
Y. Dumont, J. M. Tchuenche, Mathematical studies on the sterile insect technique for the Chikungunya disease and Aedes albopictus, J. Math. Biol., 65 (2012), 809–854. https://doi.org/10.1007/s00285-011-0477-6 doi: 10.1007/s00285-011-0477-6
|
| [17] |
Y. Dumont, I. V. Yatat-Djeument, Sterile insect technique with accidental releases of sterile females. Impact on mosquito-borne diseases control when viruses are circulating, Math. Biosci., 343 (2022), 108724. https://doi.org/10.1016/j.mbs.2021.108724 doi: 10.1016/j.mbs.2021.108724
|
| [18] |
L. Almeida, M. Duprez, Y. Privat, N. Vauchelet, Mosquito population control strategies for fighting against arboviruses, Math. Biosci. Eng., 16 (2019), 6274–6297. https://doi.org/10.3934/mbe.2019313 doi: 10.3934/mbe.2019313
|
| [19] |
S. Ai, M. Fox, Four positive equilibria in a model for sterile and wild mosquito populations, Appl. Math. Lett., 121 (2021), 107409. https://doi.org/10.1016/j.aml.2021.107409 doi: 10.1016/j.aml.2021.107409
|
| [20] |
S. Xue, M. Li, J. Ma, J. Li, Sex-structured wild and sterile mosquito population models with different release strategies, Math. Biosci. Eng., 16 (2019), 1313–1333. https://doi.org/10.3934/mbe.2019064 doi: 10.3934/mbe.2019064
|
| [21] |
S. S. Lee, R. E. Baker, E. A. Gaffney, S. M. White, Modelling Aedes Aegypti mosquito control via transgenic and sterile insect techniques Endemics and emerging outbreaks, J. Theor. Biol., 331 (2013), 78–90. https://doi.org/10.1016/j.jtbi.2013.04.014 doi: 10.1016/j.jtbi.2013.04.014
|
| [22] |
R. Anguelov, Y. Dumont, J. Lubuma, Mathematical modeling of sterile insect technology for control of anopheles mosquito, Comput. Math. Appl., 64 (2012), 374–389. https://doi.org/10.1016/j.camwa.2012.02.068 doi: 10.1016/j.camwa.2012.02.068
|
| [23] |
M. Huang, X. Song, J. Li, Modelling and analysis of impulsive releases of sterile mosquitoes, J. Biol. Dyn., 11 (2017), 147–171. https://doi.org/10.1080/17513758.2016.1254286 doi: 10.1080/17513758.2016.1254286
|
| [24] |
X. Zhang, S. Tang, R. A. Cheke, Birth-pulse models of Wolbachia-induced cytoplasmic incompatibility in mosquitoes for dengue virus control, Nonlinear Anal. Real World Appl., 22 (2015), 236–258. https://doi.org/10.1016/j.nonrwa.2014.09.004 doi: 10.1016/j.nonrwa.2014.09.004
|
| [25] |
Y. Li, X. Liu, An impulsive model for Wolbachia infection control of mosquito-borne diseases with general birth and death rate functions, Nonlinear Anal. Real World Appl., 37 (2017), 412–432. https://doi.org/10.1016/j.nonrwa.2017.03.003 doi: 10.1016/j.nonrwa.2017.03.003
|
| [26] |
P. A. Bliman, D. C. Salgadob, Y. Dumont, O. Vasilieva, Implementation of control strategies for sterile insect techniques, Math. Biosci., 314 (2019), 43–60. https://doi.org/10.1016/j.mbs.2019.06.002 doi: 10.1016/j.mbs.2019.06.002
|
| [27] | V. A. Dyck, J. Hendrichs, A. S. Robinson, The Sterile Insect Technique, Principles and Practice in Area-wide Integrated Pest Management, Springer, Dordrecht, 2006. https://doi.org/10.1201/9781003035572 |
| [28] |
B. Zheng, J. Yu, J. Li, Modeling and analysis of the implementation of the Wolbachia incompatible and sterile insect technique for mosquito population suppression, SIAM J. Appl. Math., 81 (2021), 718–740. https://doi.org/10.1137/20M1368367 doi: 10.1137/20M1368367
|
| [29] |
X. Zheng, D. Zhang, Y. Li, S. M. White, Incompatible and sterile insect techniques combined eliminate mosquitoes, Nature, 572 (2019), 56–61. https://doi.org/10.1038/s41586-019-1407-9 doi: 10.1038/s41586-019-1407-9
|
| [30] |
D. O. Carvalho, J. A. Torres-Monzon, P. Koskinioti, N. D. A. D. Wijegunawardana, X. Liang, G. Pillwax, et al., Aedes Aegypti lines for combined sterile insect technique and incompatible insect technique applications: the importance of host genomic background, Entomol. Exp. Appl., 168 (2020), 560–572. https://doi.org/10.1111/eea.12892 doi: 10.1111/eea.12892
|
| [31] |
X. Xu, Y. Xiao, R. A. Cheke, Models of impulsive culling of mosquitoes to interrupt transmission of West Nile virus to birds, Appl. Math. Modell., 39 (2015), 3549–3568. https://doi.org/10.1016/j.apm.2014.10.072 doi: 10.1016/j.apm.2014.10.072
|
| [32] |
Y. Li, X. Liu, A sex-structured model with birth pulse and release strategy for the spread of Wolbachia in mosquito population, J. Theor. Biol., 448 (2018), 53–65. https://doi.org/10.1016/j.jtbi.2018.04.001 doi: 10.1016/j.jtbi.2018.04.001
|
| [33] | H. L. Smith, Monotone Dynamical Systems: An Introduction to the Theory of Competitive and Cooperative Systems (Mathematical Surveys And Monographs), American Mathematical Society, 1995. https://doi.org/10.1090/surv/041 |
Figures(5)

Rajivganthi Chinnathambi, Fathalla A. Rihan. Analysis and control of Aedes Aegypti mosquitoes using sterile-insect techniques with Wolbachia[J]. Mathematical Biosciences and Engineering, 2022, 19(11): 11154-11171. doi: 10.3934/mbe.2022520







 DownLoad:
DownLoad: