Citation: Nick Henscheid. Generating patient-specific virtual tumor populations with reaction-diffusion models and molecular imaging data[J]. Mathematical Biosciences and Engineering, 2020, 17(6): 6531-6556. doi: 10.3934/mbe.2020341

| [1] |
R. C. Rockne, A. Hawkins-Daarud, K. R. Swanson, J. P. Sluka, J. A. Glazier, P. Macklin, et al., The 2019 mathematical oncology roadmap, Phys. Biol., 16 (2019), 041005. doi: 10.1088/1478-3975/ab1a09

|
| [2] |
J. T. Oden, E. A. Lima, R. C. Almeida, Y. Feng, M. N. Rylander, D. Fuentes, et al., Toward predictive multiscale modeling of vascular tumor growth, Arch. Comput. Methods Eng., 23 (2016), 735-779. doi: 10.1007/s11831-015-9156-x
|
| [3] | T. E. Yankeelov, N. Atuegwu, D. Hormuth, J. A. Weis, S. L. Barnes, M. I. Miga, et al., Clinically relevant modeling of tumor growth and treatment response, Sci. Trans. Med., 5 (2013), 187ps9. |
| [4] | A. Baldock, R. Rockne, A. Boone, M. Neal, A. Hawkins-Daarud, D. Corwin, et al., From patientspecific mathematical neuro-oncology to precision medicine, Front. Oncol., 3 (2013). |
| [5] | N. Henscheid, E. Clarkson, K. J. Myers, H. H. Barrett, Physiological random processes in precision cancer therapy, PloS One, 13 (2018), e0199823. |
| [6] | N. Henscheid, Quantifying Uncertainties in Imaging-Based Precision Medicine, Ph.D thesis, University of Arizona, 2018. |
| [7] |
P. L. Bedard, A. R. Hansen, M. J. Ratain, L. L. Siu, Tumour heterogeneity in the clinic, Nature, 501 (2013), 355. doi: 10.1038/nature12627
|
| [8] |
R. Everett, K. B. Flores, N. Henscheid, J. Lagergren, K. Larripa, D. Li, et al., A tutorial review of mathematical techniques for quantifying tumor heterogeneity, Math. Biosci. Eng., 17 (2020), 3660. doi: 10.3934/mbe.2020207
|
| [9] | H. Barrett, K. Myers, Foundations of Image Science, John Wiley & Sons, New York, 2004. |
| [10] | K. Swanson, R. Rostomily, E. Alvord Jr, A mathematical modelling tool for predicting survival of individual patients following resection of glioblastoma: a proof of principle, Br. J. Cancer, 98 (2008), 113. |
| [11] | A. Badano, C. G. Graff, A. Badal, D. Sharma, R. Zeng, F. W. Samuelson, et al., Evaluation of digital breast tomosynthesis as replacement of full-field digital mammography using an in silico imaging trial, JAMA Network Open, 1 (2018), e185474. |
| [12] | A. Karolak, D. A. Markov, L. J. McCawley, K. A. Rejniak, Towards personalized computational oncology: from spatial models of tumour spheroids, to organoids, to tissues, J. R. Soc., Interface, 15 (2018), 20170703. |
| [13] | E. Abadi, W. P. Segars, B. M. Tsui, P. E. Kinahan, N. Bottenus, A. F. Frangi, et al., Virtual clinical trials in medical imaging: a review, J. Med. Imaging, 7 (2020), 042805. |
| [14] |
K. R. Swanson, C. Bridge, J. Murray, E. C. Alvord, Virtual and real brain tumors: using mathematical modeling to quantify glioma growth and invasion, J. Neurol. Sci., 216 (2003), 1-10. doi: 10.1016/j.jns.2003.06.001
|
| [15] |
P. Tracqui, Biophysical models of tumour growth, Rep. Prog. Phys., 72 (2009), 056701. doi: 10.1088/0034-4885/72/5/056701
|
| [16] | J. T. Oden, E. E. Prudencio, A. Hawkins-Daarud, Selection and assessment of phenomenological models of tumor growth, Math. Models Methods Appl. Sci., 23 (2013), 1309-1338. |
| [17] |
E. M. Rutter, T. L. Stepien, B. J. Anderies, J. D. Plasencia, E. C. Woolf, A. C. Scheck, et al., Mathematical analysis of glioma growth in a murine model, Sci. Rep., 7 (2017), 2508. doi: 10.1038/s41598-017-02462-0
|
| [18] |
J. A. Weis, M. I. Miga, L. R. Arlinghaus, X. Li, A. B. Chakravarthy, V. Abramson, et al., A mechanically coupled reaction-diffusion model for predicting the response of breast tumors to neoadjuvant chemotherapy, Phys. Med. Biol., 58 (2013), 5851. doi: 10.1088/0031-9155/58/17/5851
|
| [19] |
C. Hogea, C. Davatzikos, G. Biros, An image-driven parameter estimation problem for a reaction-diffusion glioma growth model with mass effects, J. Math. Biol., 56 (2008), 793-825. doi: 10.1007/s00285-007-0139-x
|
| [20] |
T. L. Stepien, E. M. Rutter, Y. Kuang, Traveling waves of a go-or-grow model of glioma growth, SIAM J. Appl. Math., 78 (2018), 1778-1801. doi: 10.1137/17M1146257
|
| [21] |
E. J. Kostelich, Y. Kuang, J. M. McDaniel, N. Z. Moore, N. L. Martirosyan, M. C. Preul, Accurate state estimation from uncertain data and models: an application of data assimilation to mathematical models of human brain tumors, Biol. Direct, 6 (2011), 64. doi: 10.1186/1745-6150-6-64
|
| [22] | M. Gyllenberg, G. F. Webb, Quiescence as an explanation of Gompertzian tumor growth, Growth Dev. Aging., 53 (1989), 25-33. |
| [23] | J. D. Logan, An Introduction to Nonlinear Partial Differential Equations, John Wiley & Sons, 2008. |
| [24] | W. Hundsdorfer, J. G. Verwer, Numerical Solution of Time-Dependent Advection-DiffusionReaction Equations, Springer Science & Business Media, 2013. |
| [25] | R. C. Smith, Uncertainty Quantification: Theory, Implementation, and Applications, SIAM, 2013. |
| [26] | X. Han, P. E. Kloeden, Random Ordinary Differential Equations and their Numerical Solution, Springer, 2017. |
| [27] | G. J. Lord, C. E. Powell, T. Shardlow, Introduction to Computational Stochastic PDEs, Cambridge, 2014. |
| [28] |
E. Clarkson, H. H. Barrett, Characteristic functionals in imaging and image-quality assessment: tutorial, JOSA A, 33 (2016), 1464-1475. doi: 10.1364/JOSAA.33.001464
|
| [29] | I. Gel'fand, N. Vilenkin, Generalized Functions, Vol. 4: Applications of Harmonic Analysis, Academic Press, New York, 1964. |
| [30] | T. Sullivan, Introduction to Uncertainty Quantification, Springer, 2015. |
| [31] |
J. Rolland, H. H. Barrett, Effect of random background inhomogeneity on observer detection performance, JOSA A, 9 (1992), 649-658. doi: 10.1364/JOSAA.9.000649
|
| [32] |
F. Bochud, C. Abbey, M. Eckstein, Statistical texture synthesis of mammographic images with clustered lumpy backgrounds, Opt. Express, 4 (1999), 33-42. doi: 10.1364/OE.4.000033
|
| [33] |
L. G. Kessler, H. X. Barnhart, A. J. Buckler, K. R. Choudhury, M. V. Kondratovich, A. Toledano, et al., The emerging science of quantitative imaging biomarkers terminology and definitions for scientific studies and regulatory submissions, Stat. Methods Med. Res., 24 (2015), 9-26. doi: 10.1177/0962280214537333
|
| [34] |
J. P. O'connor, E. O. Aboagye, J. E. Adams, H. J. Aerts, S. F. Barrington, A. J. Beer, et al., Imaging biomarker roadmap for cancer studies, Nat. Rev. Clin. Oncol., 14 (2017), 169. doi: 10.1038/nrclinonc.2016.162
|
| [35] |
A. M. Jarrett, D. Faghihi, D. A. H. II, E. A. Lima, J. Virostko, G. Biros, et al., Optimal control theory for personalized therapeutic regimens in oncology: Background, history, challenges, and opportunities, J. Clin. Med., 9 (2020), 1314. doi: 10.3390/jcm9051314
|
| [36] |
A. Anderson, J. Xie, J. Pizzonia, R. Bronen, D. Spencer, J. Gore, Effects of cell volume fraction changes on apparent diffusion in human cells, Magn. Reson. Imaging, 18 (2000), 689-695. doi: 10.1016/S0730-725X(00)00147-8
|
| [37] |
D. A. Hormuth II, J. A. Weis, S. L. Barnes, M. I. Miga, E. C. Rericha, V. Quaranta, et al., Predicting in vivo glioma growth with the reaction diffusion equation constrained by quantitative magnetic resonance imaging data, Phys. Biol., 12 (2015), 046006. doi: 10.1088/1478-3975/12/4/046006
|
| [38] | J. Czernin, Oncological applications of FDG-PET, in PET Molecular Imaging and its Biological Applications, Springer, (2004), 321-388. |
| [39] |
R. M. Hoffman, Application of GFP imaging in cancer, Lab. Invest., 95 (2015), 432-452. doi: 10.1038/labinvest.2014.154
|
| [40] | R. Schafer, H. M. Leung, A. F. Gmitro, Multi-modality imaging of a murine mammary window chamber for breast cancer research, BioTechniques, 57 (2014), 45-50. |
| [41] | H. Vesselle, J. Grierson, M. Muzi, J. M. Pugsley, R. A. Schmidt, P. Rabinowitz, et al., In vivo validation of 3 deoxy-3-[18F] fluorothymidine ([18F] FLT) as a proliferation imaging tracer in humans: correlation of [18F] FLT uptake by positron emission tomography with KI-67 immunohistochemistry and flow cytometry in human lung tumors, Clin. Cancer Res., 8 (2002), 3315-3323. |
| [42] | E. T. McKinley, G. D. Ayers, R. A. Smith, S. A. Saleh, P. Zhao, M. K., et al., Limits of [18 F]-FLT PET as a biomarker of proliferation in oncology, PloS One, 8 (2013), e58938. |
| [43] |
E. Clarkson, M. A. Kupinski, H. H. Barrett, L. Furenlid, A task-based approach to adaptive and multimodality imaging, Proc. IEEE, 96 (2008), 500-511. doi: 10.1109/JPROC.2007.913553
|
| [44] |
H. H. Barrett, D. W. Wilson, B. M. Tsui, Noise properties of the EM algorithm. I. theory, Phys. Med. Biol., 39 (1994), 833. doi: 10.1088/0031-9155/39/5/004
|
| [45] |
Y. Ding, L. Caucci, H. H. Barrett, Charged-particle emission tomography, Med. Phys., 44 (2017), 2478-2489. doi: 10.1002/mp.12245
|
| [46] |
H. H. Barrett, W. C. Hunter, B. W. Miller, S. K. Moore, Y. Chen, L. R. Furenlid, Maximumlikelihood methods for processing signals from gamma-ray detectors, IEEE Trans. Nuclear Sci., 56 (2009), 725-735. doi: 10.1109/TNS.2009.2015308
|
| [47] |
A. K. Jha, H. H. Barrett, E. C. Frey, E. Clarkson, L. Caucci, M. A. Kupinski, Singular value decomposition for photon-processing nuclear imaging systems and applications for reconstruction and computing null functions, Phys. Med. Biol., 60 (2015), 7359. doi: 10.1088/0031-9155/60/18/7359
|
| [48] | A. P. Dempster, N. M. Laird, D. B. Rubin, Maximum likelihood from incomplete data via the EM algorithm, J. R. Stat. Soc. Ser. B (methodol.), 39 (1977), 1-22. |
| [49] | G. McLachlan, T. Krishnan, The EM Algorithm and Extensions, John Wiley & Sons, 2007. |
| [50] |
R. H. Byrd, J. C. Gilbert, J. Nocedal, A trust region method based on interior point techniques for nonlinear programming, Math. Program., 89 (2000), 149-185. doi: 10.1007/PL00011391
|
| [51] | H. H. Barrett, K. J. Myers, C. Hoeschen, M. A. Kupinski, M. P. Little, Task-based measures of image quality and their relation to radiation dose and patient risk, Phys. Med. Biol., 60 (2015), R1. |
| [52] | H. H. Barrett, J. Denny, R. F. Wagner, K. J. Myers, Objective assessment of image quality. II. Fisher information, Fourier crosstalk, and figures of merit for task performance, JOSA A, 12 (1995), 834-852. |
| [53] | Y. Pawitan, In All Likelihood: Statistical Modelling and Inference Using Likelihood, Oxford University Press, 2001. |
| [54] |
M. Lê, H. Delingette, J. Kalpathy-Cramer, E. R. Gerstner, T. Batchelor, J. Unkelbach, et al., MRI based Bayesian personalization of a tumor growth model, IEEE Trans. Med. Imaging, 35 (2016), 2329-2339. doi: 10.1109/TMI.2016.2561098
|
| [55] |
A. M. Stuart, Inverse problems: a Bayesian perspective, Acta Numerica, 19 (2010), 451-559. doi: 10.1017/S0962492910000061
|
| [56] | J. Kaipio, E. Somersalo, Statistical and Computational Inverse Problems, Springer Science & Business Media, 2006. |
| [57] |
M. A. Kupinski, J. W. Hoppin, E. Clarkson, H. H. Barrett, Ideal-observer computation in medical imaging with use of markov-chain monte carlo techniques, JOSA A, 20 (2003), 430-438. doi: 10.1364/JOSAA.20.000430
|
| [58] |
R. Allen, T. R. Rieger, C. J. Musante, Efficient generation and selection of virtual populations in quantitative systems pharmacology models, CPT: Pharmacometrics Syst. Pharmacol., 5 (2016), 140-146. doi: 10.1002/psp4.12063
|
| [59] |
T. R. Rieger, R. J. Allen, L. Bystricky, Y. Chen, G. W. Colopy, Y. Cui, et al., Improving the generation and selection of virtual populations in quantitative systems pharmacology models, Prog. Biophys. Mol. Biol., 139 (2018), 15-22. doi: 10.1016/j.pbiomolbio.2018.06.002
|
| [60] | B. Galerne, A. Leclaire, L. Moisan, Texton noise, in Computer Graphics Forum, Wiley Online Library, 2017. |
| [61] |
M. A. Kupinski, E. Clarkson, J. W. Hoppin, L. Chen, H. H. Barrett, Experimental determination of object statistics from noisy images, JOSA A, 20 (2003), 421-429. doi: 10.1364/JOSAA.20.000421
|
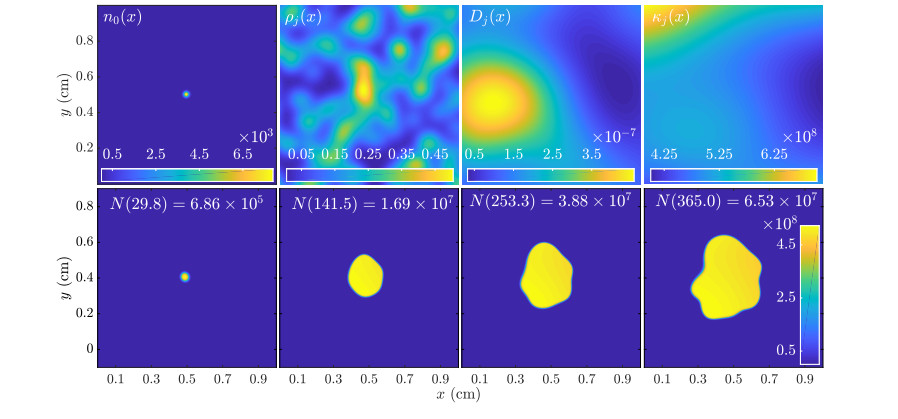
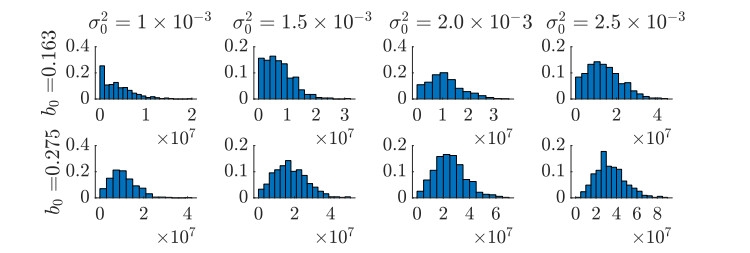
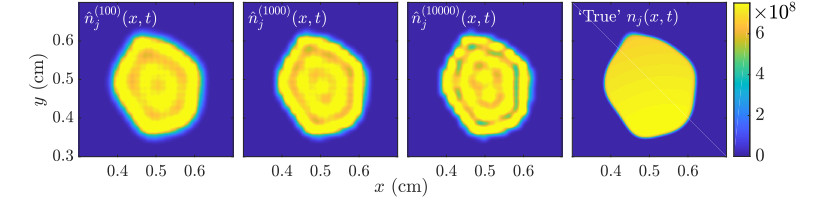
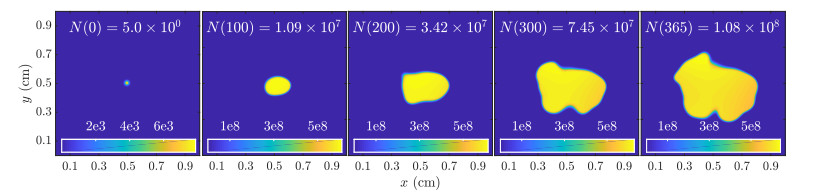
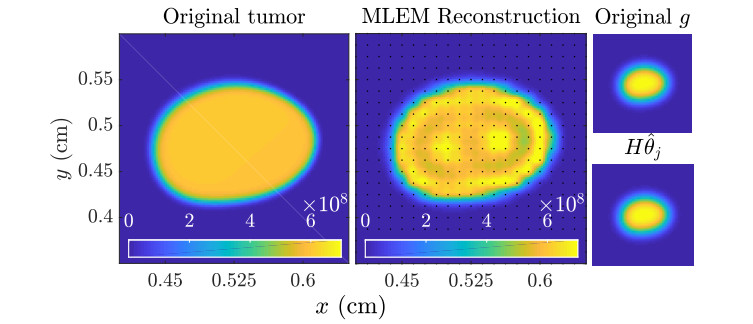
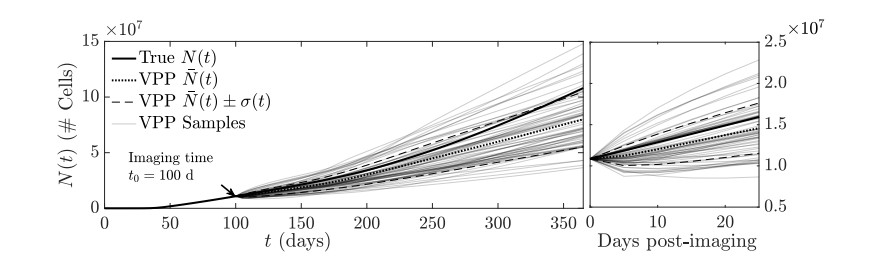
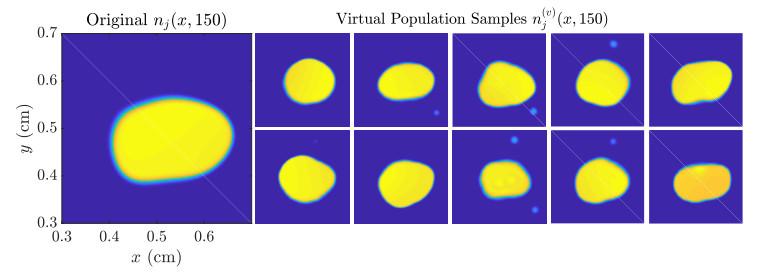
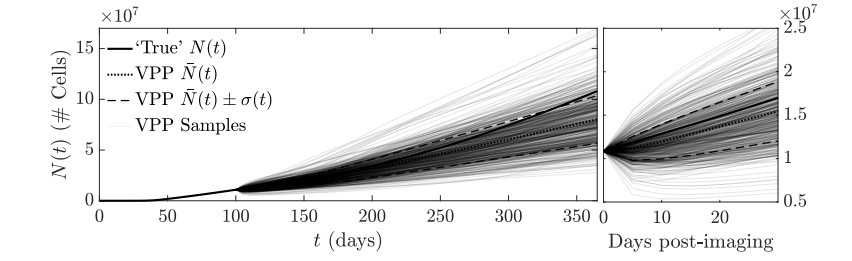
Figures(11)

Nick Henscheid. Generating patient-specific virtual tumor populations with reaction-diffusion models and molecular imaging data[J]. Mathematical Biosciences and Engineering, 2020, 17(6): 6531-6556. doi: 10.3934/mbe.2020341







 DownLoad:
DownLoad: