Citation: Anne Heponiemi, Said Azalim, Tao Hu, Tuomas Vielma, Ulla Lassi. Efficient removal of bisphenol A from wastewaters: Catalytic wet air oxidation with Pt catalysts supported on Ce and Ce–Ti mixed oxides[J]. AIMS Materials Science, 2019, 6(1): 25-44. doi: 10.3934/matersci.2019.1.25

| [1] | Corrales J, Kristofco LA, Steele WB, et al. (2015) Global assessment of bisphenol A in the environment: Review and analysis of its occurrence and bioaccumulation. Dose-Response 13: 1559325815598308. |
| [2] |
Meeker JD, Calafat AM, Hauser R (2010) Urinary bisphenol A concentrations in relation to serum thyroid and reproductive hormone levels in men from an infertility clinic. Environ Sci Technol 44: 1458–1463. doi: 10.1021/es9028292

|
| [3] | Hassan ZK, Elobeid MA, Virk P, et al. (2012) Bisphenol A induces hepatotoxicity through oxidative stress in rat model. Oxid Med Cell Longev 2012: 194829. |
| [4] |
Helmestam M, Davey E, Stavreus-Evers A, et al. (2014) Bisphenol A affects human endometrial endothelial cell angiogenic activity in vitro. Reprod Toxicol 46: 69–76. doi: 10.1016/j.reprotox.2014.03.002
|
| [5] |
Li Y, Jin F, Wang C, et al. (2015) Modification of bentonite with cationic surfactant for the enhanced retention of bisphenol A from landfill leachate. Environ Sci Pollut R 22: 8618–8628. doi: 10.1007/s11356-014-4068-0
|
| [6] |
Rocha S, Domingues V, Pinho C, et al. (2013) Occurrence of bisphenol A, estrone, 17β-estradiol and 17α-ethinylestradiol in Portuguese Rivers. B Environ Contam Tox 90: 73–78. doi: 10.1007/s00128-012-0887-1
|
| [7] | Lee CC, Jiang LY, Kuo YL, et al. (2013) The potential role of water quality parameters on occurrence of nonylphenol and bisphenol A and identification of their discharge sources in the river ecosystems. Chemosphere 91: 904–911. |
| [8] |
Kawagoshi Y, Fujita Y, Kishi I, et al. (2003) Estrogenic chemicals and estrogenic activity in leachate from municipal waste landfill determined by yeast two-hybrid assay. J Environ Monitor 5: 269–274. doi: 10.1039/b210962j
|
| [9] |
Coors A, Jones P, Giesy J, et al. (2003) Removal of estrogenic activity from municipal waste landfill leachate assessed with a bioassay based on reporter gene expression. Environ Sci Technol 37: 3430–3434. doi: 10.1021/es0300158
|
| [10] |
Lee H, Peart TE, Chan J, et al. (2004) Occurrence of endocrine-disrupting chemicals in sewage and sludge samples in Toronto, Canada. Water Qual Res J Can 39: 57–63. doi: 10.2166/wqrj.2004.009
|
| [11] | Hoigné J, Bader H, Haag WR, et al. (1985) Rate constants of reactions of ozone with organic and inorganic compounds in water-III. Inorganic compounds and radicals. Water Res 19: 993–1004. |
| [12] | Spivack J, Leib TK, Lobos JH (1994) Novel pathway for bacterial metabolism of bisphenol A. Rearrangements and stilbene cleavage in bisphenol A metabolism. J Biol Chem 269: 7323–7329. |
| [13] |
Marttinen SK, Kettunen RH, Rintala JA (2003) Occurrence and removal of organic pollutants in sewages and landfill leachates. Sci Total Environ 301: 1–12. doi: 10.1016/S0048-9697(02)00302-9
|
| [14] |
Clara M, Strenn B, Saracevic E, et al. (2004) Adsorption of bisphenol-A, 17β-estradiole and 17α-ethinylestradiole to sewage sludge. Chemosphere 56: 843–851. doi: 10.1016/j.chemosphere.2004.04.048
|
| [15] | Kondrakov AO, Ignatev AN, Frimmel FH, et al. (2014) Formation of genotoxic quinones during bisphenol A degradation by TiO2 photocatalysis and UV photolysis: A comparative study. Appl Catal B-Environ 160: 106–114. |
| [16] |
Richard J, Boergers A, vom Eyser C, et al. (2014) Toxicity of the micropollutants bisphenol A, ciprofloxacin, metoprolol and sulfamethoxazole in water samples before and after the oxidative treatment. Int J Hyg Envir Heal 217: 506–514. doi: 10.1016/j.ijheh.2013.09.007
|
| [17] |
Juhola R, Heponiemi A, Tuomikoski S, et al. (2017) Preparation of novel Fe catalysts from industrial by-products: Catalytic wet peroxide oxidation of bisphenol A. Top Catal 60: 1387–1400. doi: 10.1007/s11244-017-0829-6
|
| [18] | Erjavec B, Kaplan R, Djinovic P, et al. (2013) Catalytic wet air oxidation of bisphenol A model solution in a trickle-bed reactor over titanate nanotube-based catalysts. Appl Catal B-Environ 132–133: 342–352. |
| [19] | Levec J, Pintar A (2007) Catalytic wet-air oxidation processes: A review. Catal Today 124: 172–184. |
| [20] | Luck F (1999) Wet air oxidation: Past, present and future. Catal Today 53: 81–91. |
| [21] | Sassi H, Lafaye G, Amor HB, et al. (2017) Wastewater treatment by catalytic wet air oxidation process over Al–Fe pillared clays synthesized using microwave irradiation. Front Env Sci Eng 12: 2–7. |
| [22] |
De Los Monteros AE, Lafaye G, Cervantes A, et al. (2015) Catalytic wet air oxidation of phenol over metal catalyst (Ru, Pt) supported on TiO2–CeO2 oxides. Catal Today 258: 564–569. doi: 10.1016/j.cattod.2015.01.009
|
| [23] |
Zhang Y, Zhou Y, Peng C, et al. (2018) Enhanced activity and stability of copper oxide/γ-alumina catalyst in catalytic wet-air oxidation: Critical roles of cerium incorporation. Appl Surf Sci 436: 981–988. doi: 10.1016/j.apsusc.2017.12.036
|
| [24] |
Schmit F, Bois L, Chassagneux F, et al. (2015) Catalytic wet air oxidation of methylamine over supported manganese dioxide catalysts. Catal Today 258: 570–575. doi: 10.1016/j.cattod.2014.12.034
|
| [25] |
Yang S, Zhu W, Wang J, et al. (2008) Catalytic wet air oxidation of phenol over CeO2–TiO2 catalyst in the batch reactor and the packed-bed reactor. J Hazard Mater 153: 1248–1253. doi: 10.1016/j.jhazmat.2007.09.084
|
| [26] |
Yang S, Zhu W, Jiang Z, et al. (2006) The surface properties and the activities in catalytic wet air oxidation over CeO2–TiO2 catalysts. Appl Surf Sci 252: 8499–8505. doi: 10.1016/j.apsusc.2005.11.067
|
| [27] |
Saroha AK (2017) Treatment of industrial organic raffinate containing pyridine and its derivatives by coupling of catalytic wet air oxidation and biological processes. J Clean Prod 162: 973–981. doi: 10.1016/j.jclepro.2017.06.066
|
| [28] |
Yadav A, Verma N (2018) Carbon bead-supported copper-dispersed carbon nanofibers: An efficient catalyst for wet air oxidation of industrial wastewater in a recycle flow reactor. J Ind Eng Chem 67: 448–460. doi: 10.1016/j.jiec.2018.07.019
|
| [29] |
Kim K, Ihm S (2011) Heterogeneous catalytic wet air oxidation of refractory organic pollutants in industrial wastewaters: A review. J Hazard Mater 186: 16–34. doi: 10.1016/j.jhazmat.2010.11.011
|
| [30] |
Gaálová J, Barbier J, Rossignol S (2010) Ruthenium versus platinum on cerium materials in wet air oxidation of acetic acid. J Hazard Mater 181: 633–639. doi: 10.1016/j.jhazmat.2010.05.059
|
| [31] |
Wang J, Zhu W, He X, et al. (2008) Catalytic wet air oxidation of acetic acid over different ruthenium catalysts. Catal Commun 9: 2163–2167. doi: 10.1016/j.catcom.2008.04.019
|
| [32] |
Azalim S, Franco M, Brahmi R, et al. (2011) Removal of oxygenated volatile organic compounds by catalytic oxidation over Zr–Ce–Mn catalysts. J Hazard Mater 188: 422–427. doi: 10.1016/j.jhazmat.2011.01.135
|
| [33] |
Kolaczkowski ST, Plucinski P, Beltran FJ, et al. (1999) Wet air oxidation: A review of process technologies and aspects in reactor design. Chem Eng J 73: 143–160. doi: 10.1016/S1385-8947(99)00022-4
|
| [34] | International Centre for Diffraction Data (ICDD) (2013) PDF-4+ powder diffraction database. 12 Campus Boulevard Newton Square, PA 19073-3273, USA. |
| [35] |
El Fallah J, Hilaire L, Roméo M, et al. (1995) Effect of surface treatments, photon and electron impacts on the ceria 3d core level. J Electron Spectrosc 73: 89–103. doi: 10.1016/0368-2048(94)02266-6
|
| [36] | Park PW, Ledford JS (1996) Effect of crystallinity on the photoreduction of cerium oxide: A study of CeO2 and Ce/Al2O3 catalysts. Langmuir 12: 1794–1799. |
| [37] |
Zhao B, Shi B, Zhang X, et al. (2011) Catalytic wet hydrogen peroxide oxidation of H-acid in aqueous solution with TiO2–CeO2 and Fe/TiO2–CeO2 catalysts. Desalination 268: 55–59. doi: 10.1016/j.desal.2010.09.050
|
| [38] |
Zhang XH, Luo LT, Duan ZH (2005) Preparation and application of Ce-doped mesoporous TiO2 oxide. React Kinet Catal Lett 87: 43–50. doi: 10.1007/s11144-006-0007-5
|
| [39] |
Francisco MSP, Mastelaro VR, Nascente PAP, et al. (2001) Activity and characterization by XPS, HR-TEM, raman spectroscopy, and BET surface area of CuO/CeO2–TiO2 catalysts. J Phys Chem B 105: 10515–10522. doi: 10.1021/jp0109675
|
| [40] |
Dipti SS, Chung UC, Chung WS (2007) Characteristics of the carbon nanotubes supported Pt–Ni and Ni electrocatalysts for DMFC. Met Mater Int 13: 257–260. doi: 10.1007/BF03027814
|
| [41] |
Luo N, Fu X, Cao F, et al. (2008) Glycerol aqueous phase reforming for hydrogen generation over Pt catalyst-Effect of catalyst composition and reaction conditions. Fuel 87: 3483–3489. doi: 10.1016/j.fuel.2008.06.021
|
| [42] |
Shyu JZ, Weber WH, Gandhi HS (1988) Surface characterization of alumina-supported ceria. J Phys Chem 92: 4964–4970. doi: 10.1021/j100328a029
|
| [43] | Laachir A, Perrichon V, Badri A, et al. (1991) Reduction of CeO2 by hydrogen. Magnetic susceptibility and Fourier-transform infrared, ultraviolet and X-ray photoelectron spectroscopy measurements. J Chem Soc Faraday Trans 87: 1601–1609. |
| [44] | Galtayries A, Sporken R, Riga J, et al. (1998) XPS comparative study of ceria/zirconia mixed oxides: Powders and thin film characterisation. J Electron Spectrosc 88–91: 951–956. |
| [45] |
Dauscher A, Hilaire L, Le Normand F, et al. (1990) Characterization by XPS and XAS of supported Pt/TiO2–CeO2 catalysts. Surf Interface Anal 16: 341–346. doi: 10.1002/sia.740160173
|
| [46] |
Larsson PO, Andersson A (1998) Complete oxidation of CO, ethanol, and ethyl acetate over copper oxide supported on titania and ceria modified titania. J Catal 179: 72–89. doi: 10.1006/jcat.1998.2198
|
| [47] |
Larachi F, Pierre J, Adnot A, et al. (2002) Ce 3d XPS study of composite CexMn1−xO2−y wet oxidation catalysts. Appl Surf Sci 195: 236–250. doi: 10.1016/S0169-4332(02)00559-7
|
| [48] | Alifanti M, Baps B, Blangenois N, et al. (2003) Characterization of CeO2–ZrO2 mixed oxides. comparison of the citrate and sol–gel preparation methods. Chem Mater 15: 395–403. |
| [49] |
Bedrane S, Descorme C, Duprez D (2002) Investigation of the oxygen storage process on ceria- and ceria–zirconia-supported catalysts. Catal Today 75: 401–405. doi: 10.1016/S0920-5861(02)00089-5
|
| [50] |
Bera P, Priolkar KR, Gayen A, et al. (2003) Ionic dispersion of Pt over CeO2 by the combustion method: Structural investigation by XRD, TEM, XPS, and EXAFS. Chem Mater 15: 2049–2060. doi: 10.1021/cm0204775
|
| [51] |
Tiernan MJ, Finlayson OE (1998) Effects of ceria on the combustion activity and surface properties of Pt/Al2O3 catalysts. Appl Catal B-Environ 19: 23–35. doi: 10.1016/S0926-3373(98)00055-1
|
| [52] |
Hori CE, Permana H, Ng KYS, et al. (1998) Thermal stability of oxygen storage properties in a mixed CeO2–ZrO2 system. Appl Catal B-Environ 16: 105–117. doi: 10.1016/S0926-3373(97)00060-X
|
| [53] |
Ohko Y, Ando I, Niwa C, et al. (2001) Degradation of bisphenol A in water by TiO2 photocatalyst. Environ Sci Technol 35: 2365–2368. doi: 10.1021/es001757t
|
| [54] |
Mezohegyi G, Erjavec B, Kaplan R, et al. (2013) Removal of bisphenol A and its oxidation products from aqueous solutions by sequential catalytic wet air oxidation and biodegradation. Ind Eng Chem Res 52: 9301–9307. doi: 10.1021/ie400998t
|
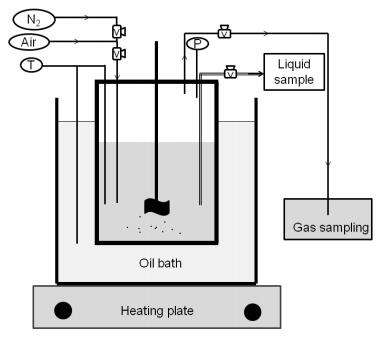
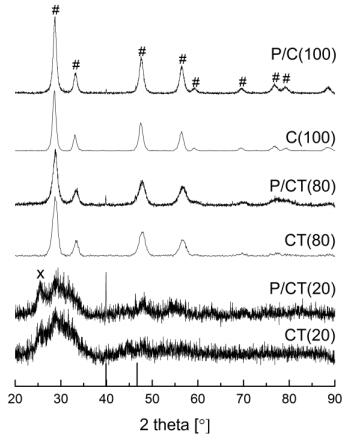
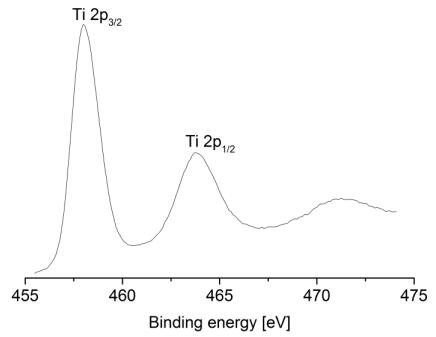
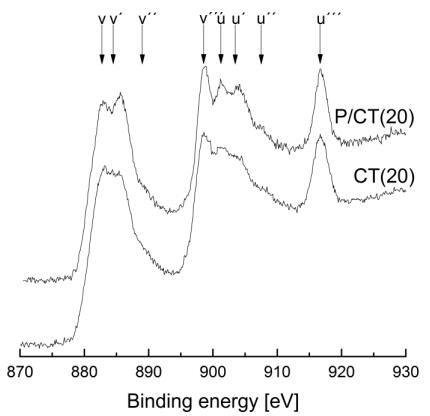
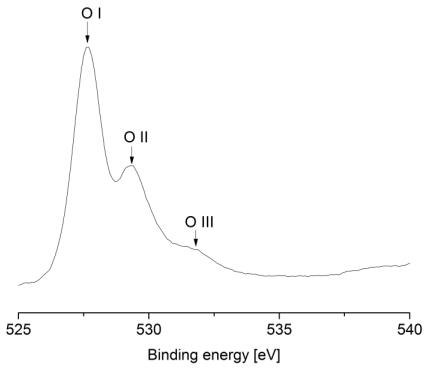

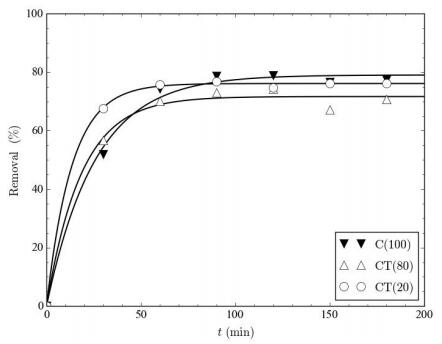
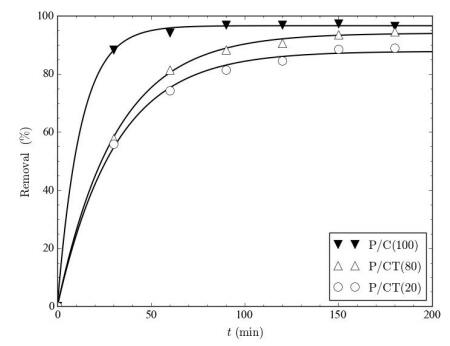
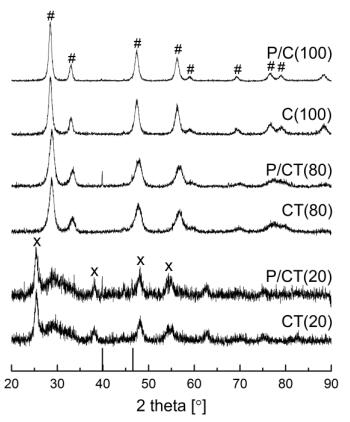
Figures(9) / Tables(9)

Anne Heponiemi, Said Azalim, Tao Hu, Tuomas Vielma, Ulla Lassi. Efficient removal of bisphenol A from wastewaters: Catalytic wet air oxidation with Pt catalysts supported on Ce and Ce–Ti mixed oxides[J]. AIMS Materials Science, 2019, 6(1): 25-44. doi: 10.3934/matersci.2019.1.25







 DownLoad:
DownLoad: