Citation: Suman Chhetri, Pranab Samanta, Naresh Chandra Murmu, Suneel Kumar Srivastava, Tapas Kuila. Electromagnetic interference shielding and thermal properties of non-covalently functionalized reduced graphene oxide/epoxy composites[J]. AIMS Materials Science, 2017, 4(1): 61-74. doi: 10.3934/matersci.2017.1.61

| [1] |
Panigrahi R, Srivastava SK (2015) Trapping of Microwave Radiation in Hollow Polypyrrole Microsphere through Enhanced Internal Reflection: A Novel Approach. Sci Rep 5: 7638. doi: 10.1038/srep07638

|
| [2] |
Kathirgamanathan P (1993) Novel cable shielding materials based on the impregnation of microporous membranes with inherently conducting polymers. Adv Mater 5: 281–283. doi: 10.1002/adma.19930050412
|
| [3] |
Li P, Du D, Guo L, et al. (2016) Stretchable and conductive polymer films for high-performance electromagnetic interference shielding. J Mater Chem C 4: 6525. doi: 10.1039/C6TC01619G
|
| [4] | Chhetri S, Kuila T, Murmu NC (2016) Graphene Composites, In: Nazarpour S, Waite SR, Eds, Graphene Technology: From Laboratory to Fabrication, John Wiley & Sons, 63–102. |
| [5] |
Chen Z, Xu C, Ma C, et al. (2013) Lightweight and Flexible Graphene Foam Composites for High-Performance Electromagnetic Interference Shielding. Adv Mater 25: 1296–1300. doi: 10.1002/adma.201204196
|
| [6] |
Li N, Huang Y, Du F, et al. (2006) Electromagnetic Interference (EMI) Shielding of Single-Walled Carbon Nanotube Epoxy Composites. Nano Lett 6: 1141–1145. doi: 10.1021/nl0602589
|
| [7] | Yang S, Lozano K, Lomeli A, et al. (2005) Electromagnetic interference shielding effectiveness of carbon nanofiber/LCP composites. Compos Part A-Appl S 36: 691–697. |
| [8] |
Farukh M, Dhawan R, Singh BP (2015) Sandwich composites of polyurethane reinforced with poly(3,4-ethylene dioxythiophene)-coated multiwalled carbon nanotubes with exceptional electromagnetic interference shielding properties. RSC Adv 5: 75229–75238. doi: 10.1039/C5RA14105B
|
| [9] |
Ling J, Zhai W, Feng W, et al. (2013) Facile Preparation of Lightweight Microcellular Polyetherimide/Graphene Composite Foams for Electromagnetic Interference Shielding. ACS Appl Mater Inter 5: 2677–2684. doi: 10.1021/am303289m
|
| [10] |
Chen Y, Zhang HB, Yang Y, et al. (2016) High-Performance Epoxy Nanocomposites Reinforced with Three-Dimensional Carbon Nanotube Sponge for Electromagnetic Interference Shielding. Adv Funct Mater 26: 447–455. doi: 10.1002/adfm.201503782
|
| [11] |
Maiti S, Shrivastava NK, Suin S, et al. (2013) Polystyrene/MWCNT/Graphite Nanoplate Nanocomposites: Efficient Electromagnetic Interference Shielding Material through Graphite Nanoplate–MWCNT–Graphite Nanoplate Networking. ACS Appl Mater Inter 5: 4712–4724. doi: 10.1021/am400658h
|
| [12] |
Liang J, Wang Y, Huang Y, et al. (2009) Electromagnetic interference shielding of graphene/epoxy composites. Carbon 47: 922–925. doi: 10.1016/j.carbon.2008.12.038
|
| [13] |
Singh BP, Choudhary V, Saini P, et al. (2012) Designing of epoxy composites reinforced with carbon nanotubes grown carbon fiber fabric for improved electromagnetic interference shielding. AIP Adv 2: 022151. doi: 10.1063/1.4730043
|
| [14] |
Lonkar SP, Deshmukh YS, Abdala AA, et al. (2015) Recent advances in chemical modifications of graphene. Nano Res 8: 1039–1074. doi: 10.1007/s12274-014-0622-9
|
| [15] |
Kuila T, Bose S, Mishra AK, et al. (2012) Chemical functionalization of graphene and its applications. Prog Mater Sci 57: 1061–1105. doi: 10.1016/j.pmatsci.2012.03.002
|
| [16] |
Jana M, Saha S, Khanra P, et al. (2015) Non-covalent functionalization of reduced graphene oxide using sulfanilic acid azocromotrop and its application as supercapacitor electrode material. J Mater Chem A 3: 7323–7331. doi: 10.1039/C4TA07009G
|
| [17] | Du J, Cheng HM (2012) The fabrication, properties, and uses of graphene/polymer composites. Macromol Chem Phys 213: 1060−1077. |
| [18] | Liang J, Huang Y, Zhang L, et al. (2009) Molecular-Level Dispersion of Graphene into Poly(vinyl alcohol) and Effective Reinforcement of their Nanocomposites. Adv Funct Mater 19: 2297−2302. |
| [19] | Wang Y, Shi ZX, Fang JH, et al. (2011) Graphene oxide/polybenzimidazole composites fabricated by a solvent-exchange method. Carbon 49: 1199−1207. |
| [20] | Zaman I, Kuan HC, Meng QS, et al. (2012) A Facile Approach to Chemically Modified Graphene and its Polymer Nanocomposites. Adv Funct Mater 22: 2735−2743. |
| [21] | Zaman I, Kuan HC, Dai JF, et al. (2012) From carbon nanotubes and silicate layers to graphene platelets for polymer nanocomposites. Nanoscale 4: 4578−4586. |
| [22] |
Tang LC, Wan YJ, Yan D, et al. (2013) The effect of graphene dispersion on the mechanical properties of graphene/epoxy composites. Carbon 60: 16–27. doi: 10.1016/j.carbon.2013.03.050
|
| [23] |
Verma P, Saini P, Malik RS, et al. (2015) Excellent electromagnetic interference shielding and mechanical properties of high loading carbon-nanotubes/polymer composites designed using melt recirculation equipped twin-screw extruder. Carbon 89: 308–317. doi: 10.1016/j.carbon.2015.03.063
|
| [24] |
Wang JC, Xiang CS, Liu Q, et al. (2008) Ordered Mesoporous Carbon/Fused Silica Composites. Adv Funct Mater 18: 2995–3002. doi: 10.1002/adfm.200701406
|
| [25] | Zhang HB, Yan Q, Zheng WG, et al. (2011) Tough Graphene-Polymer Microcellular Foams for Electromagnetic Interference Shielding. ACS Appl Mater Inter 3: 918−924. |
| [26] | Fang M, Zhen Z, Li J, et al. (2010) Constructing hierarchically structured interphases for strong and tough epoxy nanocomposites by amine-rich graphene surfaces. J Mater Chem 20: 9635−9643. |
| [27] | Chhetri S, Samanta P, Murmu NC, et al. (2016) Effect of Dodecyal Amine Functionalized Graphene on the Mechanical and Thermal Properties of Epoxy-based Composites. Polym Eng Sci [In Press]. |
| [28] | Jin FL, Ma CJ, Park SJ, et al. (2011) Thermal and mechanical interfacial properties of epoxy composites based on functionalized carbon nanotubes. Mater Sci Eng A 528: 8517−8522. |
| [29] | Yu G, Wu P (2014) Effect of chemically modified graphene oxide on the phase separation behaviour and properties of an epoxy/polyetherimide binary system. Polym Chem 5: 96−104. |
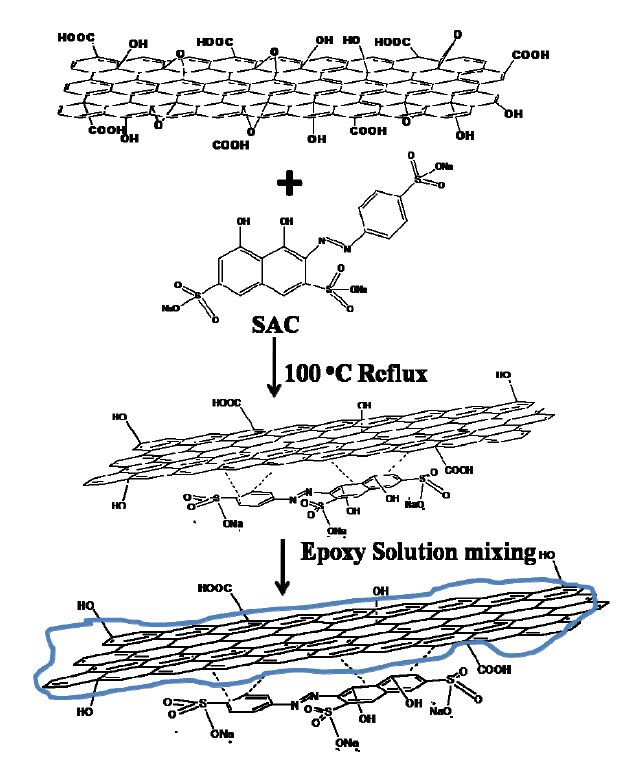
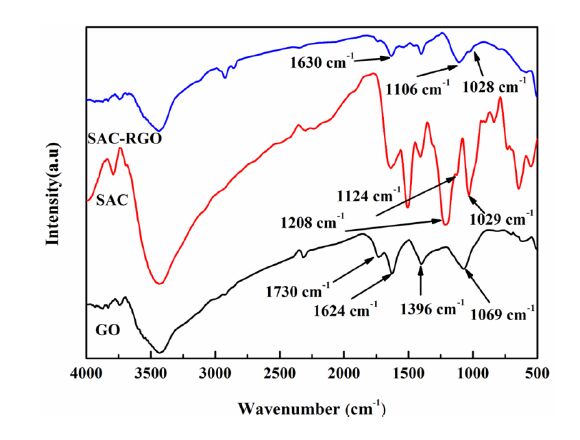

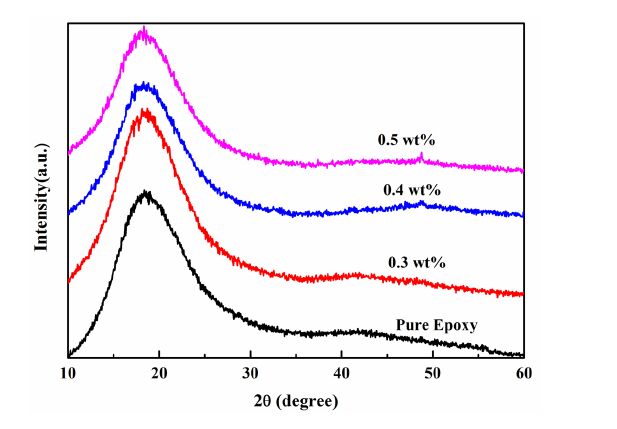

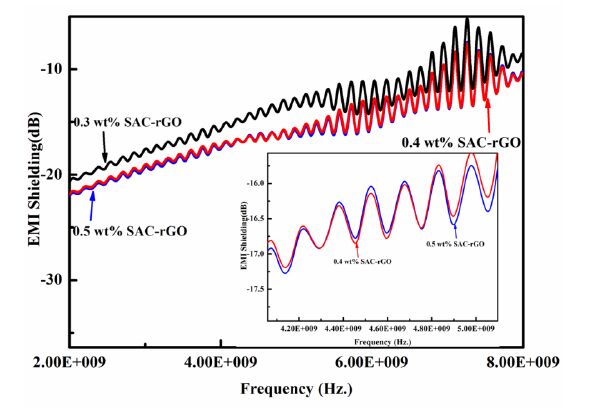
Figures(9) / Tables(1)

Suman Chhetri, Pranab Samanta, Naresh Chandra Murmu, Suneel Kumar Srivastava, Tapas Kuila. Electromagnetic interference shielding and thermal properties of non-covalently functionalized reduced graphene oxide/epoxy composites[J]. AIMS Materials Science, 2017, 4(1): 61-74. doi: 10.3934/matersci.2017.1.61







 DownLoad:
DownLoad: