Citation: Lu Wang, Chris M. Marin, Wai-Ning Mei, Chin Li Cheung. Electronic structures of lanthanum, samarium, and gadolinium sulfides[J]. AIMS Materials Science, 2015, 2(2): 97-105. doi: 10.3934/matersci.2015.2.97

| [1] |
Parida B, Iniyan S, Goic R (2011) A review of solar photovoltaic technologies. Renew Sust Energy Rev 15: 1625-1636. doi: 10.1016/j.rser.2010.11.032

|
| [2] |
Dughaish ZH (2002) Lead telluride as a thermoelectric material for thermoelectric power generation. Physica B 322: 205-223. doi: 10.1016/S0921-4526(02)01187-0
|
| [3] |
Jeon H, Ding J, Patterson W, et al. (1991) Blue-green injection laser diodes in (Zn, Cd)Se/ZnSe quantum wells. Appl Phys Lett 59: 3619-3621. doi: 10.1063/1.105625
|
| [4] |
Hardman R (2006) A toxicologic review of quantum dots: Toxicity depends on physicochemical and environmental factors. Environ Health Perspect 114: 165-172. doi: 10.1289/ehp.8284
|
| [5] | Gschneidner KA, Beaudry BJ (1995) Rare Earth Compounds, In: Rowe DM, CRC Handbook for Thermoelectrics, CRC Press. |
| [6] | Meyer G, Morss LR (1991) Synthesis of lanthanide and actinide compounds, Norwell: Kluwer Academic Publishers. |
| [7] |
Batlogg B, Kaldis E, Schlegel A, et al. (1976) Electronic structure of Sm monochalcogenides. Phys Rev B 14: 5503-5514. doi: 10.1103/PhysRevB.14.5503
|
| [8] |
Fairchild S, Cahay M, Murray PT, et al. (2012) Grain size, texture, and crystallinity in lanthanum monosulfide thin films grown by pulsed laser deposition. Thin Solid Films 524: 166-172. doi: 10.1016/j.tsf.2012.10.022
|
| [9] |
Witz C, Huhuenin D, Lafait J, et al. (1996) Comparative optical studies of Ce2S3 and Gd2S3 compounds. J Appl Phys 79: 2038-2042. doi: 10.1063/1.361058
|
| [10] |
Segall MD, Philip JDL, Probert MJ, et al. (2002) First-principles simulation: ideas, illustrations and the CASTEP code. Phys Condens Matter 14: 2717-2744. doi: 10.1088/0953-8984/14/11/301
|
| [11] |
Holtzberg F, Methfessel S (1966) Rare-Earth Compounds with the Th3P4-Type Structure. J Appl Phys 37: 1433-1435. doi: 10.1063/1.1708500
|
| [12] |
Perdew JP, Burke K, Ernzerhof M (1996) Generalized gradient approximation made simple. Phys Rev Lett 77: 3865-3868. doi: 10.1103/PhysRevLett.77.3865
|
| [13] | Petersen M, Hafner J, Marsman M (2006) Structural, electronic and magnetic properties of Gd investigated by DFT+U methods: bulk, clean and H-covered (0001) surfaces. J Phy—Condens Mat 18: 7021-7043. |
| [14] | Jeffries JR, Veiga LSI, Fabbris G, et al. (2014) Robust ferromagnetism in the compressed permanent magnet Sm2Co17. Physical Review B 90. |
| [15] |
Marzari N, Vanderbilt D, Payne MC (1997) Ensemble density functional theory for ab initio molecular dynamics of metals and finite-temperature insulators. Phys Rev Lett 79: 1337-1340. doi: 10.1103/PhysRevLett.79.1337
|
| [16] |
Payne MC, Teter MP, Allan DC, et al. (1992) Iterative minimization techniques for ab initio total-energy calculations: Molecular dynamics and conjugate gradients. Rev Mod Phys 64: 1045-1097. doi: 10.1103/RevModPhys.64.1045
|
| [17] |
Eriksson O, Wills J, Mumford P, et al. (1998) Electronic structure of the LaS surface and LaS/CdS interface. Phys Rev B 57: 4067-4072. doi: 10.1103/PhysRevB.57.4067
|
| [18] | Cotton FA, Wilkinson G (1966) Advanced inorganic chemistry; second ed.; Interscience Publishers: New York. |
| [19] | Shim JH, Kim K, Min BI, et al. (2002) Electronic structures of La3S4 and Ce3S4. Physica B 328:148-150. |
| [20] | Cahay M, Boolchand P, Fairchild SB, et al. (2011) Review article: Rare-earth monosulfides as durable and efficient cold cathodes. J Vac Sci Technol B 29: 602-1-14. |
| [21] | Strange P (1985) A band theory description of the valence transition in samarium sulphide. Physica 130B: 44-46. |
| [22] |
Batlogg B, Kaldis E, Schlegel A, et al. (1976) Optical and electrical properties of the mixed valence compound Sm3S4. Solid State Commun 19: 673-676. doi: 10.1016/0038-1098(76)91102-9
|
| [23] |
Marin CM, Wang L, Brewer JR, et al. (2013) Crystalline α-Sm2S3 nanowires: Structure and optical properties of an unusual intrinsically degenerate semiconductor. J Alloys Compd 563:293-299. doi: 10.1016/j.jallcom.2013.02.082
|
| [24] | Grazhulis VA, Bozhko SI, Bolotin IL, et al. (1996) Electronic structure of the valence band of GdSx and Gd3S4. Appl Surf Sci 104/105: 68-72. |
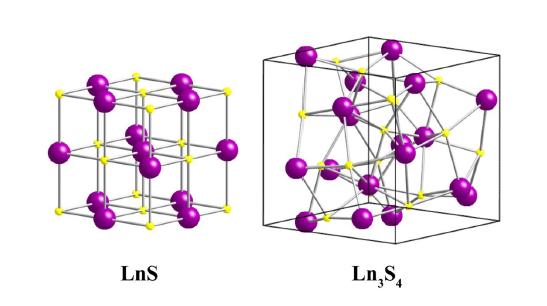
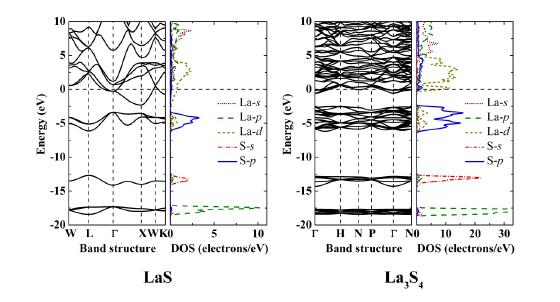
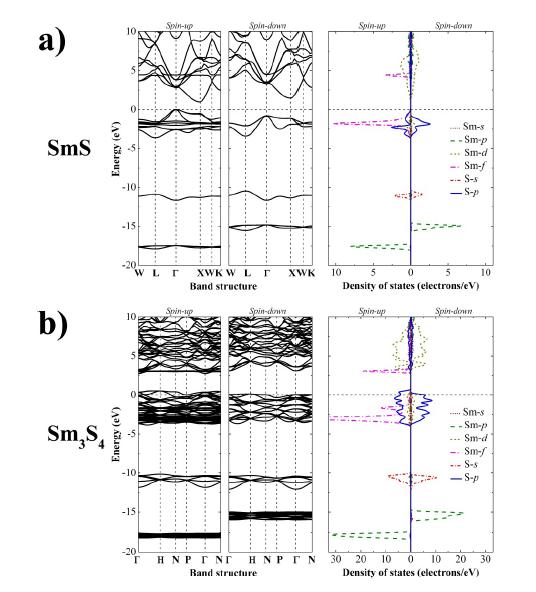
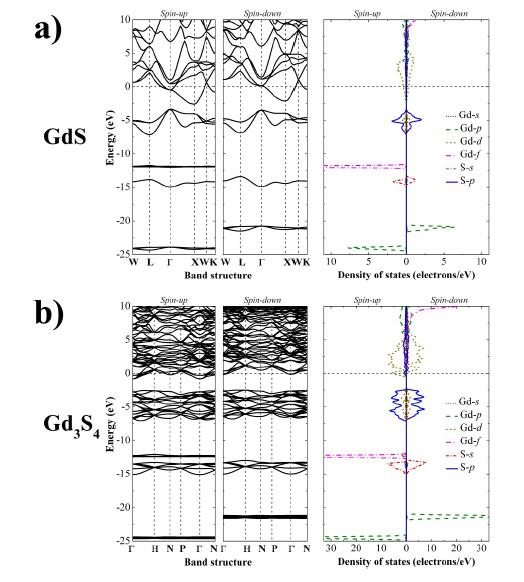
Figures(4)

Lu Wang, Chris M. Marin, Wai-Ning Mei, Chin Li Cheung. Electronic structures of lanthanum, samarium, and gadolinium sulfides[J]. AIMS Materials Science, 2015, 2(2): 97-105. doi: 10.3934/matersci.2015.2.97







 DownLoad:
DownLoad: