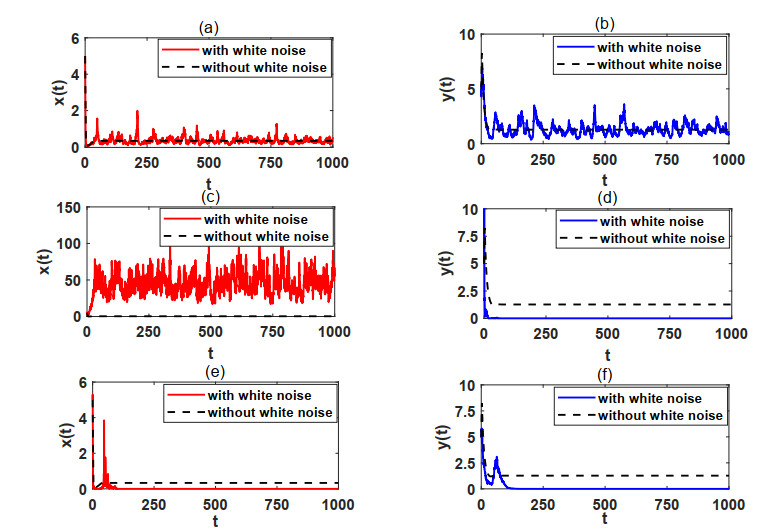
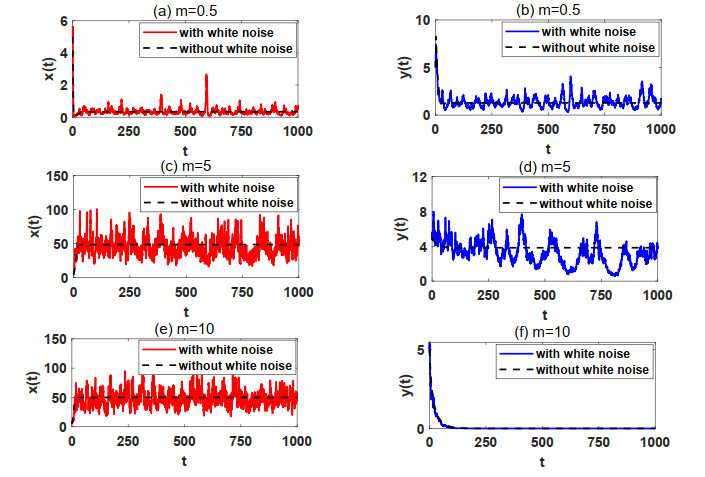
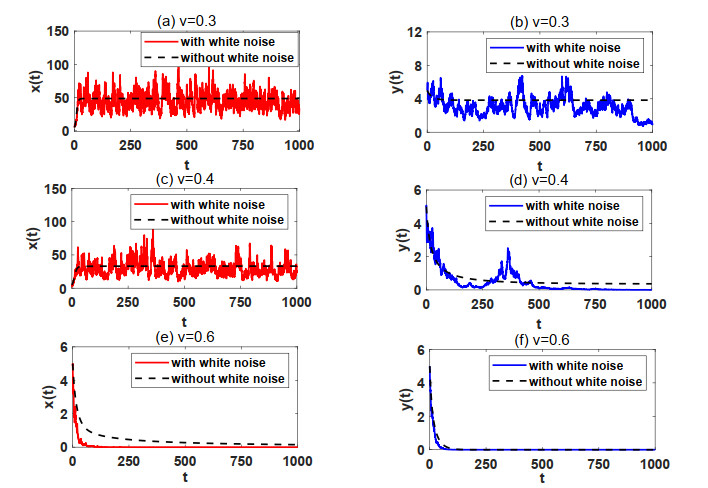
In natural ecosystems, the external environment is constantly changing, and is affected by various factors, thus presenting a certain degree of randomness and uncertainty. Therefore, having a suitable habitat is essential for the reproductive success of many species. Understanding the impact of habitat selection provides valuable insights into how species locate and adapt to suitable living environments based on their specific needs. For this purpose, a prey-predator system model with prey habitat selection in an environment subject to stochastic disturbances is formulated. The properties of the proposed model without and with stochastic disturbances are investigated, including the existence of a unique ergodic stationary distribution, the stochastically ultimate bounded-ness of the solutions, and the extinction and persistence of the populations. The study demonstrates that prey can persist at a low intensity noise, whereas stronger stochastic disturbances may lead to the extinction of both the prey and predator species. To illustrate the theoretical results, numerical simulations are presented step by step. This work provides a theoretical reference for further studies on populations with habitat selection in an environment subject to stochastic disturbances.
Citation: Yuan Tian, Jing Zhu, Jie Zheng, Kaibiao Sun. Modeling and analysis of a prey-predator system with prey habitat selection in an environment subject to stochastic disturbances[J]. Electronic Research Archive, 2025, 33(2): 744-767. doi: 10.3934/era.2025034
In natural ecosystems, the external environment is constantly changing, and is affected by various factors, thus presenting a certain degree of randomness and uncertainty. Therefore, having a suitable habitat is essential for the reproductive success of many species. Understanding the impact of habitat selection provides valuable insights into how species locate and adapt to suitable living environments based on their specific needs. For this purpose, a prey-predator system model with prey habitat selection in an environment subject to stochastic disturbances is formulated. The properties of the proposed model without and with stochastic disturbances are investigated, including the existence of a unique ergodic stationary distribution, the stochastically ultimate bounded-ness of the solutions, and the extinction and persistence of the populations. The study demonstrates that prey can persist at a low intensity noise, whereas stronger stochastic disturbances may lead to the extinction of both the prey and predator species. To illustrate the theoretical results, numerical simulations are presented step by step. This work provides a theoretical reference for further studies on populations with habitat selection in an environment subject to stochastic disturbances.

| [1] |
Y. Kuang, E. Beretta, Global qualitative analysis of a ratio-dependent predator-prey system, J. Math. Biol., 36 (1998), 389–406. https://doi.org/10.1007/s002850050105 doi: 10.1007/s002850050105

|
| [2] |
D. Xiao, S. Ruan, Global analysis in a predator-prey system with nonmonotonic functional response, SIAM J. Appl. Math., 61 (2001), 1445–1472. https://doi.org/10.1137/S0036139999361896 doi: 10.1137/S0036139999361896
|
| [3] |
T. Zheng, L. Nie, Modelling the transmission dynamics of two-strain Dengue in the presence awareness and vector control, J. Theor. Biol., 443 (2018), 82–91. https://doi.org/10.1016/j.jtbi.2018.01.017 doi: 10.1016/j.jtbi.2018.01.017
|
| [4] |
L. Nie, J. Shen, C. Yang. Dynamic behavior analysis of SIVS epidemic models with state-dependent pulse vaccination, Nonlinear Anal: Hybrid Syst., 27 (2018), 258–270. https://doi.org/10.1016/j.nahs.2017.08.004 doi: 10.1016/j.nahs.2017.08.004
|
| [5] |
X. Yan, Y. Tian, K. Sun, Dynamic analysis of additional food provided non-smooth pest-natural enemy models based on nonlinear threshold control, J. Appl. Math. Comput., (2025). https://doi.org/10.1007/s12190-024-02318-7 doi: 10.1007/s12190-024-02318-7
|
| [6] | A. J. Lotka, Elements of Physical Biology, Williams and Wilkins Company, Baltimore, 1925. |
| [7] |
V. Volterra, Fluctuations in the abundance of a species considered mathematically, Nature, 119 (1927), 12–13. https://doi.org/10.1038/119012b0 doi: 10.1038/119012b0
|
| [8] |
C. S. Holling, The functional response of predators to prey density and its role in mimicry and population regulation, Mem. Entomol. Soc. Can., 97 (1965), 5–60. https://doi.org/10.4039/entm9745fv doi: 10.4039/entm9745fv
|
| [9] |
M. P. Hassell, G. C. Varley, New inductive population model for insect parasites and its bearing on biological control, Nature, 223 (1969), 1133–1177. https://doi.org/10.1038/2231133a0 doi: 10.1038/2231133a0
|
| [10] |
J. R. Beddington, Mutual interference between parasites or predator and its effect on searching efficiency, J. Anim Ecol., 44 (1975), 331–340. https://doi.org/10.2307/3866 doi: 10.2307/3866
|
| [11] |
R. Arditi, L. R. Ginzburg, Coupling in predator-prey dynamics: Ratio-dependence, J. Theor. Biol., 139 (1989), 311–326. https://doi.org/10.1016/S0022-5193(89)80211-5 doi: 10.1016/S0022-5193(89)80211-5
|
| [12] |
A. Zegeling, R. E. Kooij, Singular perturbations of the Holling Ⅰ predator-prey system with a focus, J. Differ. Equations, 269 (2020), 5434–5462. https://doi.org/10.1016/j.jde.2020.04.011 doi: 10.1016/j.jde.2020.04.011
|
| [13] |
M. Lu, J. Huang, Global analysis in Bazykin's model with Holling Ⅱ functional response and predator competition, J. Differ. Equations, 280 (2021), 99–138. https://doi.org/10.1016/j.jde.2021.01.025 doi: 10.1016/j.jde.2021.01.025
|
| [14] |
J. Huang, S. Ruan, R. Song, Bifurcations in a predator-prey system of Leslie type with generalized Holling type Ⅲ functional response, J. Differ. Equations, 257 (2014), 1721–1752. https://doi.org/10.1016/j.jde.2014.04.024 doi: 10.1016/j.jde.2014.04.024
|
| [15] |
Q. Yang, X. Zhang, D. Jiang, M. Shao, Analysis of a stochastic predator-prey model with weak Allee effect and Holling-(n+1) functional response, Commun. Nonlinear Sci. Numer. Simul., 111 (2022), 106454. https://doi.org/10.1016/j.cnsns.2022.106454 doi: 10.1016/j.cnsns.2022.106454
|
| [16] |
C. B. Stanford, The influence of chimpanzee predation on group size and anti-predator behaviour in red colobus monkeys, Anim. Behav., 49 (1995), 577–587. https://doi.org/10.1016/0003-3472(95)80191-X doi: 10.1016/0003-3472(95)80191-X
|
| [17] |
M. J. How, M. Santon, Cuttlefish camouflage: Blending in by matching background features, Curr. Biol., 32 (2022), 523–525. https://doi.org/10.1016/j.cub.2022.04.042 doi: 10.1016/j.cub.2022.04.042
|
| [18] |
E. Gazagne, T. Savini, D. Ngoprasert, P. Poncin, M. Huynem, F. Brotcorne, When northern pigtailed macaques (Macaca leonina) cannot select for ideal sleeping sites in a degraded habitat, Int. J. Primatol., 41 (2020), 614–633. https://doi.org/10.1007/s10764-020-00173-4 doi: 10.1007/s10764-020-00173-4
|
| [19] |
D. Li, Q. Zhou, X. Tang, H. Huang, C. Huang, Sleeping site use of the white-headed langur Trachypithecus leucocephalus: The role of predation risk, territorial defense, and proximity to feeding sites, Curr. Zool., 57 (2011), 260–268. https://doi.org/10.1093/czoolo/57.3.260 doi: 10.1093/czoolo/57.3.260
|
| [20] |
B. Tang, Y. Xiao, Bifurcation analysis of a predator-prey model with anti-predator behaviour, Chaos Solitons Fractals, 70 (2015), 58–68. https://doi.org/10.1016/j.chaos.2014.11.008 doi: 10.1016/j.chaos.2014.11.008
|
| [21] |
Y. Tian, Y. Gao, K. Sun, A fishery predator-prey model with anti-predator behavior and complex dynamics induced by weighted fishing strategies, Math. Biosci. Eng., 20 (2023), 1558–1579. https://doi.org/10.3934/mbe.2023071 doi: 10.3934/mbe.2023071
|
| [22] |
A. R. Ives, A. P. Dobson, Antipredator behavior and the population dynamics of simple predator-prey systems, Am. Nat., 130 (1987), 431–447. https://doi.org/10.1086/284719 doi: 10.1086/284719
|
| [23] |
T. Caraco, S. Martindale, H. R. Pulliam, Avian flocking in the presence of a predator, Nature, 285 (1980), 400–401. https://doi.org/10.1038/285400a0 doi: 10.1038/285400a0
|
| [24] |
E. E. Werner, J. F. Gilliam, D. J. Hall, G. G. Mittelbach, An experimental test of the effects of predation risk on habitat use in fish, Ecology, 64 (1983), 1540–1548. https://doi.org/10.2307/1937508 doi: 10.2307/1937508
|
| [25] | R. M. May, Stability and Complexity in Model Ecosystems, Princeton university press, Princeton, 2001. https://doi.org/10.1515/9780691206912 |
| [26] |
M. Liu, K. Wang, Q. Wu, Survival analysis of stochastic competitive models in a polluted environment and stochastic competitive exclusion principle, Bull. Math. Biol., 73 (2011), 1969–2012. https://doi.org/10.1007/s11538-010-9569-5 doi: 10.1007/s11538-010-9569-5
|
| [27] |
W. Wang, W. Li, Z. Li, H. Zhang, The effect of colored noise on spatiotemporal dynamics of biological invasion in a diffusive predator-prey system, Biosystems, 104 (2011), 48–56. https://doi.org/10.1016/j.biosystems.2010.12.011 doi: 10.1016/j.biosystems.2010.12.011
|
| [28] |
G. Li, Q. Yang, Dynamics of a stochastic Holling Ⅱ predator-prey model with Lévy jumps and habitat complexity, Int. J. Biomath., 14 (2021), 2150077. https://doi.org/10.1142/S1793524521500777 doi: 10.1142/S1793524521500777
|
| [29] |
M. Li, G. Lan, C. Wei, Threshold dynamics of stochastic H7N9 model with Markov switching and hybrid strategy, J. Franklin Inst., 361 (2024), 916–932. https://doi.org/10.1016/j.jfranklin.2023.12.034 doi: 10.1016/j.jfranklin.2023.12.034
|
| [30] | L. Arnold, Stochastic Differential Equations: Theory and Applications, Wiley, New York, 1974. |
| [31] |
X. Mao, G. Marion, E. Renshaw, Environmental Brownian noise suppresses explosions in population dynamics, Stochastic Processes Appl., 97 (2002), 95–110. https://doi.org/10.1016/S0304-4149(01)00126-0 doi: 10.1016/S0304-4149(01)00126-0
|
| [32] |
C. Ji, D. Jiang, Dynamics of a stochastic density dependent predator-prey system with Beddington-DeAngelis functional response, J. Math. Anal. Appl., 381 (2011), 441–453. https://doi.org/10.1016/j.jmaa.2011.02.037 doi: 10.1016/j.jmaa.2011.02.037
|
| [33] |
Q. Liu, L. Zu, D. Jiang, Dynamics of stochastic predator-prey models with Holling Ⅱ functional response, Commun. Nonlinear Sci. Numer. Simul., 37 (2016), 62–76. https://doi.org/10.1016/j.cnsns.2016.01.005 doi: 10.1016/j.cnsns.2016.01.005
|
| [34] |
X. Zhang, Y. Li, D. Jiang, Dynamics of a stochastic Holling type Ⅱ predator-prey model with hyperbolic mortality, Nonlinear Dyn., 87 (2017), 2011–2020. https://doi.org/10.1007/s11071-016-3172-8 doi: 10.1007/s11071-016-3172-8
|
| [35] |
M. Liu, Dynamics of a stochastic regime-switching predator-prey model with modified Leslie-Gower Holling-type Ⅱ schemes and prey harvesting, Nonlinear Dyn., 96 (2019), 417–442. https://doi.org/10.1007/s11071-019-04797-x doi: 10.1007/s11071-019-04797-x
|
| [36] |
Y. Li, Z. Wei, W. Zhang, T. Kapitaniak, Melnikov-type method for chaos in a class of hybrid piecewise-smooth systems with impact and noise excitation under unilateral rigid constraint, Appl. Math. Modell., 122 (2023), 506–523. https://doi.org/10.1016/j.apm.2023.06.015 doi: 10.1016/j.apm.2023.06.015
|
| [37] | X. Mao, Stochastic Differential Equations and Applications, $2^{nd}$ edition, Woodhead Publishing, Cambridge, 2007. |
| [38] |
D. J. Higham, An algorithmic introduction to numerical simulation of stochastic differential equations, SIAM Rev., 43 (2021), 525–546. https://doi.org/10.1137/S0036144500378302 doi: 10.1137/S0036144500378302
|
Figures(7)
Yuan Tian, Jing Zhu, Jie Zheng, Kaibiao Sun. Modeling and analysis of a prey-predator system with prey habitat selection in an environment subject to stochastic disturbances[J]. Electronic Research Archive, 2025, 33(2): 744-767. doi: 10.3934/era.2025034







 DownLoad:
DownLoad: