Citation: Yu-Ting Huang, Hui-Fen Liao, Shun-Li Wang, Shan-Yang Lin. Glycation and secondary conformational changes of human serum albumin: study of the FTIR spectroscopic curve-fitting technique[J]. AIMS Biophysics, 2016, 3(2): 247-260. doi: 10.3934/biophy.2016.2.247

| [1] |
Vanhooren V,Navarrete Santos A,Voutetakis K, et al. (2015) Protein modification and maintenance systems as biomarkers of ageing. Mech Ageing Dev 151: 71–84. doi: 10.1016/j.mad.2015.03.009

|
| [2] |
Uribarri J, Woodruff S, Goodman S, et al. (2010) Advanced glycation end products in foods and a practical guide to their reduction in the diet. J Am Diet Assoc 110: 911–916.e12 doi: 10.1016/j.jada.2010.03.018
|
| [3] |
Visentin S,Medana C,Barge A, et al. (2010) Microwave-assisted Maillard reactions for the preparation of advanced glycation end products (AGEs). Org Biomol Chem 8: 2473–2477. doi: 10.1039/c000789g
|
| [4] | Horvat S,Jakas A (2014) Peptide and amino acid glycation: new insights into the Maillard reaction. J Pept Sci 10: 119–137. |
| [5] |
Zhang Q,Ames JM,Smith RD, et al. (2009) A perspective on the Maillard reaction and the analysis of protein glycation by mass spectrometry: probing the pathogenesis of chronic disease. J Proteome Res 8: 754–769. doi: 10.1021/pr800858h
|
| [6] | Dar B, Dar M, Bashir S, et al. (2015) Glycosylated hemoglobin (HbA1c): A biomarker of anti-aging. Int J Biol Med Res 6: 5084–5086. |
| [7] |
Sebeková K,Somoza V (2007) Dietary advanced glycation endproducts (AGEs) and their health effects--PRO. Mol Nutr Food Res 51: 1079–1084. doi: 10.1002/mnfr.200700035
|
| [8] | Arasteh A,Farahi S,Habibi-Rezaei M, et al. (2014) Glycated albumin: an overview of the in vitro models of an in vivo potential disease marker. J Diabetes Metab Disord 13: 49. |
| [9] |
Uribarri J,del Castillo MD,de la Maza MP, et al. (2015) Dietary advanced glycation end products and their role in health and disease. Adv Nutr 6: 461–473. doi: 10.3945/an.115.008433
|
| [10] | Takahashi M (2014) Glycation of Proteins. In Glycoscience: Biology and Medicine, Endo T, Seeberger PH, Hart GW, Wong CH, Taniguchi N, eds., Springer Japan, pp. 1339–1345 |
| [11] | Nursten HE (2005) The Maillard Reaction: Chemistry, Biochemistry, and Implications. RSC. |
| [12] |
Laroque D, Inisan C, Berger C, et al. (2008) Kinetic study on the Maillard reaction: Consideration of sugar reactivity. Food Chem 111: 1032–1042 doi: 10.1016/j.foodchem.2008.05.033
|
| [13] | Sattarahmady N,Moosavi-Movahedi AA,Habibi-Rezaei M, et al. (2008) Detergency effects of nanofibrillar amyloid formation on glycation of human serum albumin. Carbohydr Res 343: 2229–2234. |
| [14] |
Monnier VM (1990) Nonenzymatic glycosylation, the Maillard reaction and the aging process. J Gerontol 45: B105–111. doi: 10.1093/geronj/45.4.B105
|
| [15] |
Wei Y,Han CS,Zhou J, et al. (2012) D-ribose in glycation and protein aggregation. Biochim Biophys Acta 1820: 488–494. doi: 10.1016/j.bbagen.2012.01.005
|
| [16] |
Monnier VM, Cerami A (1981) Nonenzymatic browning in vivo: possible process for aging of long-lived proteins. Science 211: 491–493. doi: 10.1126/science.6779377
|
| [17] |
Han C,Lu Y,Wei Y, et al. (2011) D-ribose induces cellular protein glycation and impairs mouse spatial cognition. PLoS ONE 6:e24623. doi: 10.1371/journal.pone.0024623
|
| [18] |
Syrový I (1994) Glycation of albumin: reaction with glucose, fructose, galactose, ribose or glyceraldehyde measured using four methods. J Biochem Biophys Methods 28: 115–121. doi: 10.1016/0165-022X(94)90025-6
|
| [19] |
Kong FL,Cheng W,Chen J, et al. (2011) D-Ribose glycates β(2)-microglobulin to form aggregates with high cytotoxicity through a ROS-mediated pathway. Chem Biol Interact 194: 69–78. doi: 10.1016/j.cbi.2011.08.003
|
| [20] | Khan MS,Dwivedi S,Priyadarshini M, et al. (2013) Ribosylation of bovine serum albumin induces ROS accumulation and cell death in cancer line (MCF-7). Eur Biophys J 42: 811–818. |
| [21] |
Iannuzzi C,Maritato R,Irace G, et al. (2013) Glycation accelerates fibrillization of the amyloidogenic W7FW14F apomyoglobin. PLoS ONE 8: e80768. doi: 10.1371/journal.pone.0080768
|
| [22] |
Adrover M,Mariño L,Sanchis P, et al. (2014) Mechanistic insights in glycation-induced protein aggregation. Biomacromolecules 15: 3449–3462. doi: 10.1021/bm501077j
|
| [23] | Liu J, Ru Q, Ding Y (2012) Glycation a promising method for food protein modification: Physicochemical properties and structure, a review. Food Res Int 49: 170–183. |
| [24] | Wei Y, Chen L, Chen J, et al. (2009) Rapid glycation with D-ribose induces globular amyloid-like aggregations of BSA with high cytotoxicity to SH-SY5Y cells. BMC Cell Biol 10: 10. |
| [25] | Kragh-Hansen U,Chuang VT,Otagiri M (2002) Practical aspects of the ligand-binding and enzymatic properties of human serum albumin. Biol Pharm Bull 25: 695–704. |
| [26] | Santra MK,Banerjee A,Krishnakumar SS, et al. (2004) Multiple-probe analysis of folding and unfolding pathways of human serum albumin. Evidence for a framework mechanism of folding. Eur J Biochem 271: 1789–1797. |
| [27] | Anguizola J,Matsuda R,Barnaby OS, et al. (2013) Review: Glycation of human serum albumin. Clin Chim Acta 2013; 425: 64–76. |
| [28] | Singha Roy A,Ghosh P,Dasgupta S (2015) Glycation of human serum albumin alters its binding efficacy towards the dietary polyphenols: A comparative approach. J Biomol Struct Dyn Oct 7:1–46. [In press]. |
| [29] | Peters T (1996) All about Albumin. Biochemistry, Genetics, and Medical Applications. Academic Press, San Diego, CA. |
| [30] |
Khan MW,Rasheed Z,Khan WA, et al. (2007) Biochemical, biophysical, and thermodynamic analysis of in vitro glycated human serum albumin. Biochemistry (Mosc) 72: 146–152. doi: 10.1134/S0006297907020034
|
| [31] | Yang F,Zhang Y,Liang H (2014) Interactive association of drugs binding to human serum albumin. Int J Mol Sci 15: 3580–3595. |
| [32] |
Lin SY,Wei YS,Li MJ,et al (2004). Effect of ethanol or/and captopril on the secondary structure of human serum albumin before and after protein binding. Eur J Pharm Biopharm 57: 457–464. doi: 10.1016/j.ejpb.2004.02.005
|
| [33] |
Lin SY, Wei YS, Li MJ (2004) Ethanol or/and captopril-induced precipitation and secondary conformational changes of human serum albumin. Spectrochim Acta A 60: 3107–3111. doi: 10.1016/j.saa.2004.03.001
|
| [34] |
Li MJ,Lin SY (2005) Vibrational spectroscopic studies on the disulfide formation and secondary conformational changes of captopril-HSA mixture after UV-B irradiation. Photochem Photobiol 81: 1404–1410. doi: 10.1562/2005-04-25-RN-497
|
| [35] |
Sadowska-Bartosz I,Galiniak S,Bartosz G (2014) Kinetics of glycoxidation of bovine serum albumin by glucose, fructose and ribose and its prevention by food components. Molecules 19: 18828–18849. doi: 10.3390/molecules191118828
|
| [36] |
Kosaraju SL,Weerakkody R,Augustin MA (2010) Chitosan-glucose conjugates: influence of extent of Maillard reaction on antioxidant properties. J Agric Food Chem 58: 12449–12455. doi: 10.1021/jf103484z
|
| [37] | Ajandouz EH, Tchiakpe LS, Ore FD, et al. (2001) Effects of pH on caramelization and Maillard reaction kinetics in fructose-lysine model systems. J Food Sci 66: 926–931. |
| [38] | Monacelli F, Storace D, D’Arrigo C, et al. (2013)Structural alterations of human serum albumin caused by glycative and oxidative stressors revealed by circular dichroism analysis. Int J Mol Sci 14: 10694–10709. |
| [39] |
Lee TH,Cheng WT,Lin SY (2010) Thermal stability and conformational structure of salmon calcitonin in the solid and liquid states. Biopolymers 93: 200–207. doi: 10.1002/bip.21323
|
| [40] | Ledesma-Osuna AI, Ramos-Clamont G, Vazquez-Moreno L (2008) Characterization of bovine serum albumin glycated with glucose, galactose and lactose. Acta Biochim Pol 55: 491–497. |
| [41] |
Sompong W,Meeprom A,Cheng H, et al. (2013) A comparative study of ferulic acid on different monosaccharide-mediated protein glycation and oxidative damage in bovine serum albumin. Molecules 18: 13886–13903. doi: 10.3390/molecules181113886
|
| [42] | Wu CH, Huang SM, Lin JA, et al. (2011) Inhibition of advanced glycation endproduct formation by foodstuffs. Food Funct 2: 224–234. |
| [43] |
Kato Y, Matsuda T, Kato N, et al. (1989) Maillard reaction of disaccharides with protein: suppressive effect of nonreducing end pyranoside groups on browning and protein polymerization. J Agric Food Chem 37: 1077–1081. doi: 10.1021/jf00088a057
|
| [44] | Suárez G,Rajaram R,Oronsky AL, et al.(1989). Nonenzymatic glycation of bovine serum albumin by fructose (fructation). Comparison with the Maillard reaction initiated by glucose. J Biol Chem 264: 3674–3679. |
| [45] | McPherson JD,Shilton BH,Walton DJ (1988) Role of fructose in glycation and cross-linking of proteins. Biochemistry 27: 1901–1907. |
| [46] | Siddiqui AA,Sohail A,Bhat SA, et al. (2015). Non-enzymatic glycation of almond cystatin leads to conformational changes and altered activity. Protein Pept Lett 22: 449–459. |
| [47] |
Awasthi S,Murugan NA,Saraswathi NT (2015) Advanced glycation end products modulate structure and drug binding properties of albumin. Mol Pharmaceutics 12: 3312–3322. doi: 10.1021/acs.molpharmaceut.5b00318
|
| [48] |
Bouma B,Kroon-Batenburg LM,Wu YP, et al. (2003) Glycation induces formation of amyloid cross-beta structure in albumin. J Biol Chem 278: 41810–41819. doi: 10.1074/jbc.M303925200
|
| [49] |
Khajehpour M,Dashnau JL,Vanderkooi JM (2006) Infrared spectroscopy used to evaluate glycosylation of proteins. Anal Biochem 348: 40–48. doi: 10.1016/j.ab.2005.10.009
|
| [50] |
GhoshMoulick R,Bhattacharya J,Roy S, et al. (2007). Compensatory secondary structure alterations in protein glycation. Biochim Biophys Acta 1774: 233–242. doi: 10.1016/j.bbapap.2006.11.018
|
| [51] |
Yang H,Yang S,Kong J, et al. (2015). Obtaining information about protein secondary structures in aqueous solution using Fourier transform IR spectroscopy. Nat Protoc 10: 382–396. doi: 10.1038/nprot.2015.024
|
| [52] | Roy R,Boskey A,Bonassar LJ (2010) Processing of type I collagen gels using nonenzymatic glycation. J Biomed Mater Res A 93: 843–851. |
| [53] | Haris PI (2013) Probing protein-protein interaction in biomembranes using Fourier transform infrared spectroscopy. Biochim Biophys Acta 1828: 2265–2271. |
| [54] | Neault JF, Tajmir-Riahi HA (1998) Interaction of cisplatin with human serum albumin. Drug binding mode and protein secondary structure, Biochim. Biophys Acta 1384: 153–159. |
| [55] | Bramanti E, Benedetti E (1996) Determination of the secondary structure of isomeric forms of human serum albumin by a particular frequency deconvolution procedure applied to Fourier transform IR analysis. Biopolymers 38: 639–653. |
| [56] |
Zsila F (2013) Subdomain IB is the third major drug binding region of human serum albumin: toward the three-sites model. Mol Pharmaceutics 10: 1668–1682. doi: 10.1021/mp400027q
|
| [57] | Awasthi S,Murugan NA,Saraswathi NT (2015) Advanced glycation end products modulate structure and drug binding properties of albumin. Mol Pharmaceutics 12: 3312–3322. |
| [58] |
Khan TA, Saleemuddin M, Naeem A (2011) Partially folded glycated state of human serum albumin tends to aggregate. Int J Pept Res Ther 17: 271–279. doi: 10.1007/s10989-011-9267-7
|
| [59] |
Oliveira LM,Lages A,Gomes RA, et al. (2011) Insulin glycation by methylglyoxal results in native-like aggregation and inhibition of fibril formation. BMC Biochem 5; 12:41. doi: 10.1186/1471-2091-12-41
|
| [60] | Lin SY,Chu HL,Wei YS (2002) Pressure-induced transformation of alpha-helix to beta-sheet in the secondary structures of amyloid beta (1–40) peptide exacerbated by temperature. J Biomol Struct Dyn 19: 619–625. |
| [61] |
Ding F,Borreguero JM,Buldyrey SV, et al. (2003) Mechanism for the alpha-helix to beta-hairpin transition. Proteins 53: 220–228. doi: 10.1002/prot.10468
|
| [62] |
Garip S,Yapici E,Ozek NS, et al. (2010) Evaluation and discrimination of simvastatin-induced structural alterations in proteins of different rat tissues by FTIR spectroscopy and neural network analysis. Analyst 135: 3233–3241. doi: 10.1039/c0an00540a
|
| [63] | Yano K,Ohoshima S,Shimizu Y, et al. (1996) Evaluation of glycogen level in human lung carcinoma tissues by an infrared spectroscopic method. Cancer Lett 110: 29–34. |
| [64] |
Podshyvalov A,Sahu RK,Mark S, et al. (2005) Distinction of cervical cancer biopsies by use of infrared microspectroscopy and probabilistic neural networks. Appl Opt 44: 3725–3734. doi: 10.1364/AO.44.003725
|
| [65] | Colagar AH,Chaichi MJ,Khadjvand T (2011) Fourier transform infrared microspectroscopy as a diagnostic tool for distinguishing between normal and malignant human gastric tissue. J Biosci 36: 669–677. |
| [66] |
Nagai R, Shirakawa J, Fujiwara Y, et al. (2014) Detection of AGEs as markers for carbohydrate metabolism and protein denaturation. J Clin Biochem Nutr 55: 1–6. doi: 10.3164/jcbn.13-112
|
| [67] | Rondeau P,Bourdon E (2011) The glycation of albumin: structural and functional impacts. Biochimie 93: 645–658. |
| [68] |
Basta G,Schmidt AM,De Caterina R (2004) Advanced glycation end products and vascular inflammation: implications for accelerated atherosclerosis in diabetes. Cardiovasc Res 63: 582–592. doi: 10.1016/j.cardiores.2004.05.001
|
| [69] |
Shivu B,Seshadri S,Li J, et al. (2013) Distinct β-sheet structure in protein aggregates determined by ATR-FTIR spectroscopy. Biochemistry 52: 5176–5183. doi: 10.1021/bi400625v
|
| [70] |
Natalello A,Doglia SM (2015) Insoluble protein assemblies characterized by fourier transform infrared spectroscopy. Methods Mol Biol 1258: 347–369. doi: 10.1007/978-1-4939-2205-5_20
|
| [71] | Clark AH,Saunderson DH,Suggett A (1981) Infrared and laser-Raman spectroscopic studies of thermally-induced globular protein gels. Int J Pept Protein Res 17: 353–364. |
| [72] | Ruggeri FS,Longo G,Faggiano S, et al. (2015) Infrared nanospectroscopy characterization of oligomeric and fibrillar aggregates during amyloid formation. Nat Commun 6: 7831. |
| [73] | Miller LM,Bourassa MW,Smith RJ (2013) FTIR spectroscopic imaging of protein aggregation in living cells. Biochim Biophys Acta 1828: 2339–2346. |
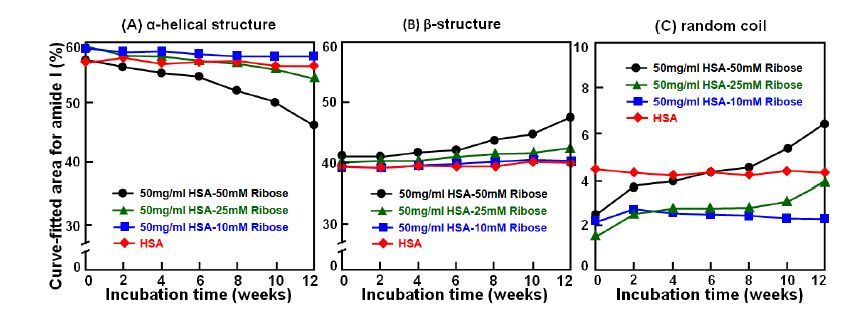
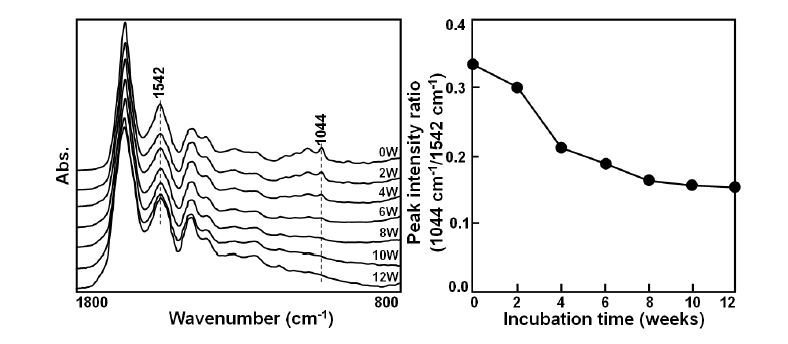
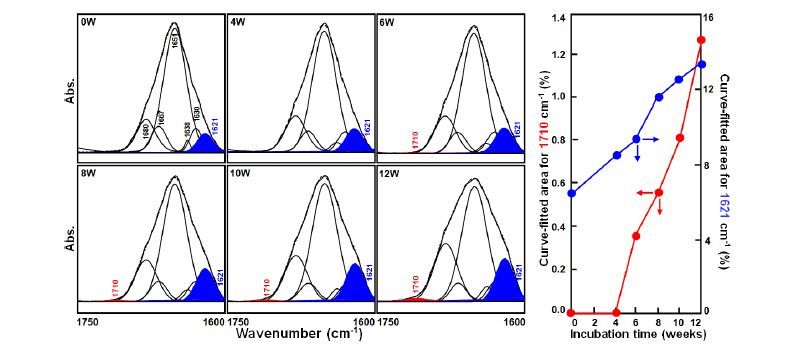
Figures(7)

Yu-Ting Huang, Hui-Fen Liao, Shun-Li Wang, Shan-Yang Lin. Glycation and secondary conformational changes of human serum albumin: study of the FTIR spectroscopic curve-fitting technique[J]. AIMS Biophysics, 2016, 3(2): 247-260. doi: 10.3934/biophy.2016.2.247







 DownLoad:
DownLoad: